CONFERENCE COVERAGE SERIES
Society for Neuoscience Annual Meeting 2018
San Diego, California
03 – 07 November 2018

CONFERENCE COVERAGE SERIES
San Diego, California
03 – 07 November 2018
The question of which Aβ species are toxic in Alzheimer’s disease is often asked, and the answer has been maddeningly elusive. At the Society for Neuroscience annual meeting in San Diego November 3–7, the lab of Dominic Walsh, Brigham and Women’s Hospital, Boston, took a fresh stab at it with new data presented by Wei Hong. The scientists once again nominated Aβ dimers as the toxic form of the peptide found in amyloid plaques. The difference this time around? Not one, but many pairs of different Aβ monomers linked by covalent bonds. These dimers are damaging to neurons. “This is the first-ever definitive proof of a toxic dimer, and the first identification of a specific cross-link that holds monomers together,” Walsh told Alzforum.
When Aβ from aqueous brain extracts is analyzed by SDS-PAGE, a common method of separating proteins on a gel, two prominent bands emerge—a 4-kDa band consisting of Aβ monomers, and a 7-kDa band of unknown origin. The monomers seem harmless to neurons in cell and slice culture, but the 7-kDa band is toxic. Walsh, now in collaboration with Kaj Blennow, Henrik Zetterberg, Erik Portelius, and Gunnar Brinkmalm, all of the University of Gothenburg, Sweden, has spent the better part of a decade trying to pin down the molecular identity of that 7-kDa band. In 2008, he reported that they were likely dimers of Aβ (Nov 2007 conference news; Jun 2008 news). However, to show that definitively, he would need to do mass spectrometry, which proved to require an impossibly large sample of aqueous oligomers isolated from human brain.
In the present study, Hong and colleagues got around this problem by using material isolated from Aβ plaques, which litter AD brains and are a rich source of Aβ. The researchers used six autopsied brains provided by Matt Frosch at Massachusetts General Hospital. Dissolving the plaques in formic acid released 4- and 7-kDa bands that resembled those found in aqueous extracts. Once again the material in the 4-kDa was harmless to neurons, but the contents of the 7-kDa band disrupted neurites and blocked long-term potentiation in neurons derived from induced pluripotent stem cells. This time around, the researchers had enough 7-kDa material in hand for mass spectrometry that would determine the structure of the neurotoxic material.
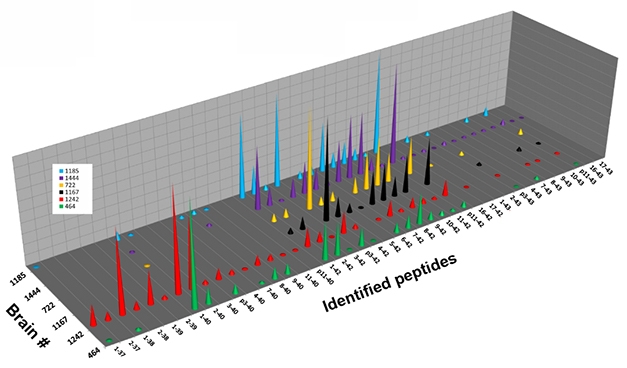
Monomer Diversity. Six individual brains (separate colors) contained different fragments of Aβ monomer, isolated from the 4-kDa band of solubilized plaques. [Image courtesy of Dominic Walsh.]
In the 4-kDa band, liquid chromatography followed by mass spectrometry revealed more than 35 different monomeric Aβ species, in different abundances in the six brains examined (see image above). Analyzing the 7-kDa band in the same way revealed a broad mix of peaks, whose masses were consistent with diverse Aβ heterodimers. The mix comprised many combinations of Aβ37 to Aβ42, the 11 most abundant of which were 1-37 plus 1-38, 1-38 plus 1-38, 1-38 plus 1-39, 1-38 plus 1-40, 1-39 plus 1-40, 1-40 plus 1-40, 2-38 plus 1-40, 2-40 plus 1-38, 2-40 plus 1-40, 1-40 plus 1-42, and 1-42 plus 1-42.

Dimer Diversity. The 7-kDa fraction of Aβ isolated from human AD plaques contains a mix of species whose component masses are consistent with Aβ dimers. [Image courtesy of Dominic Walsh.]
To determine where these dimers were cross-linked, the researchers digested the 7-kDa material with trypsin/LysC, which cleaves after arginine and lysine residues. They then used mass spec-mass spec to analyze the fragments. In addition to fragments that would be anticipated from Aβ lacking a cross-linking covalent bond, they also detected a fragment encompassing residues 1–5 and 17–28 with a cross-link between Asp1 and Glu22. That suggested the presence of dimers held together by a covalent bond.
Walsh said that so far his group has identified only one specific cross-link. Others likely exist given that many Aβ alloforms lack the Asp1 amino acid, he said. “This is likely the tip of the iceberg,” he told Alzforum.
The dimers in the 7-kDa band represent one toxic form of Aβ, though Walsh was careful to note that dimers are not the only one. Understanding which dimers are most toxic could allow researchers to target them specifically and perhaps protect neurons better. For instance, anti-Aβ antibodies that recognize covalent dimers could have more therapeutic benefit than antibodies that don’t.
The 7-kDa band isolated from the 7PA2 Chinese hamster ovary cell line, which overproduces Aβ42 due to a V717F mutation in APP, was previously reported to contain N-terminally extended fragments of Aβ, as well as large Aη fragments that are products of an alternative APP cleavage (May 2015 news; Aug 2015 news). However, in the 7-kDa fraction isolated from human brains, mass spec evidence of a covalent bond between trypsin-digested Aβ fragments, and no evidence of Aη, suggests that the toxic species in these extracts are instead dimers of Aβ, Portelius wrote to Alzforum.
“The analytical study done is exceptional and among the best mass spectrometry data to date,” wrote Randall Bateman, Washington University School of Medicine, St. Louis, to Alzforum. “It clearly identifies several novel cross-linked amyloid-β species in bands that could be oligomers.” He raised several questions, namely which of the dimers identified here are toxic in Alzheimer’s disease, and how to control for artifacts created by homogenization and extraction techniques. Bateman also wondered how these cross-linked species relate to various stages of AD, and how they cause toxicity in vivo.
“I think it’s likely that these are real, covalently linked dimers.” wrote David Brody, Uniformed Services University of the Health Sciences, Bethesda, Maryland, to Alzforum. “The big question is whether they exist in the soluble phase and at what concentration.”
Walsh plans to create antibodies specific for the Asp1-Glu22 cross-link. With them, he could determine the abundance of these dimers in native aqueous brain extracts and whether they relate to the presence and severity of the disease. He and his Swedish collaborators also want to explore whether these dimers could serve as fluid biomarkers for Alzheimer’s disease.—Gwyneth Dickey Zakaib
Never mind the wrinkles and gray hair. Embattled microglia, waning neurogenesis, and withering synapses are also part and parcel of the aging process. What drives those seemingly inexorable changes inside the brain? At the Society for Neuroscience meeting, held November 3–7 in San Diego, researchers blamed factors from the outside. Specifically, they identified circulating immune cells as key purveyors of aging in the hippocampus, culminating in memory loss in mice. One such factor, eotaxin, rises in plasma as people age. Blocking its action restored memory in aging mice, and protected mouse models of Parkinson’s disease from motor deficits, sparking early stage clinical trials in humans.
The findings extend striking previous,evidence, conducted largely by the same scientists, that systemic factors fuel age-related decline in brain function. In 2014, a trio of papers reported that blood from old mice brought on markers of brain aging in young mice, while blood from young mice ablated aging markers in old mice (May 2014 news on Villeda et al., 2014). Young blood slowed synapse loss and neuroinflammation in mice modeling Alzheimer’s disease (Sep 2016 news).
Immune Orchestra of Aging?
At SfN, researchers from the lab of Saul Villeda at the University of California, San Francisco, hypothesized that circulating immune cells were likely responsible for these parabiotic effects. To investigate whether these cells trigger aging in the brain, graduate student Luke Smith transplanted hematopoietic stem cells (HSCs) from 24-month-old mice into two-month-old mice. As a control, he transplanted other two-month-old mice with HSCs from same-age littermates. Four months later, Smith found that compared with controls, mice with old HSCs performed worse on tests of hippocampal-dependent memory, such as the water maze and fear conditioning. A look at their brains revealed less neurogenesis in the hippocampal dentate gyrus, more activated microglia, and fewer dendritic spines on hippocampal neurons than in controls.
The finding suggests that hematopoietic cells impart some of the blood’s aging influence on the brain, said Smith. Whatever the specific cell type, it likely exerts its sway by some soluble factors, he added, as nary a transplanted cell was found within the brains of recipients.
Could such factors act via microglia? After all, chronic activation of microglia is a hallmark of brain aging, and thought to modulate neurodegeneration. While exposure to old plasma can boost activation of microglia (May 2014 news), Jeremy Shea, a postdoc in Villeda’s lab, wanted to investigate whether microglia are also influenced by their local environment in the brain. Focusing on the hippocampus, Shea first simply asked how microglia there change with age. As expected, he found they became more activated, rounding up and expressing markers such as CD68. Surprisingly, this age-related activation depended on the ZIP code within the hippocampus. Microglia in the granule cell layer and hilus, but not the molecular layer, were activated.
To understand how the environment of the aging brain itself affects microglia, Shea next transplanted fledgling microglia extracted from the brains of newborn mice into the hippocampi of young or old mice. These naïve microglia assumed a more activated state after settling into old but not young hippocampi.
Returning to parabiosis, the researchers found that microglia in young mice ramped up an age-related gene expression signature after being conjoined to old mice, upregulating disease-related and immune genes. This signature was also present in hippocampal microglia from old mice, and the researchers found they could lessen it by joining old mice to youngsters. Shea said that the findings cast microglia, in keeping with their function, as exquisitely sensitive responders to both the local brain environment as well as systemic factors coming in from the blood.
Eo-taxin’ the Brain with Age
What, then, are those secret ingredients in blood that age the brain? According to Sakura Minami of the San Carlos-based biotech company Alkahest, one is eotaxin, a ligand for the C-C chemokine receptor type 3 (CCR3). Using proteomics approaches, Minami and colleagues saw that eotaxin shot up in the blood with age. Also known as CCL11, eotaxin is known to play a role in inflammation, for example in the recruitment of eosinophils upon CCR3 engagement. Given that these infection-fighting white blood cells can become damaging in allergic conditions such as asthma, CCR3 is an established drug target.
The researchers previously demonstrated that injecting eotaxin into young mice caused neurogenesis in the dentate gyrus to slow to a trickle, and that neutralizing eotaxin with an antibody blocked this effect (Aug 2011 news). At SfN, Minami reported preclinical findings from efforts to stifle the consequences of age-related eotaxin elevation. Rather than target eotaxin directly, Alkahest scientists blocked CCR3 with a small molecule antagonist acquired from Boehringer-Ingelheim. The drug, first called ALK4290, then AKST4290, was reportedly well-tolerated in previous clinical trials, though Boehringer-Ingelheim did not make results public.
First, the researchers established that rising eotaxin was detrimental to memory in mice. They put the mice through a Barnes maze test, placing them on a circular platform with holes surrounding its circumference, only one of which led to an escape box below. The mice had to use visual cues in the room to remember the escape hole. To make the test even more challenging, Minami modified the set-up to include rotating target holes. The researchers found that injecting young wild-type mice with recombinant eotaxin docked their performance on the test. Much like old mice, they were slow to locate target holes. For both sets of mice, treatment with the CCR3 antagonist corrected the deficit to the point where they performed as well as young, untreated animals, Minami reported. The researchers obtained similar results using the Y-maze, another spatial memory test.
Minami told the SfN audience that she noticed treated mice moved faster than controls through the Barnes maze, though the quickened pace did not explain their performance boost. Was there a motor benefit? Exploring this, Minami found that old mice injected with the CCR3 antagonist maintained their balance on a spinning rod nearly as long as young mice, while old control mice fell off quickly.
They next tested the drug in Line 61, Parkinson’s disease mice that overexpresses human, wild-type α-synuclein (Chesselet et al., 2012). After spiking the mice’s drinking water with the drug for six weeks, the researchers saw dramatic improvement in grip strength, and modest improvement in balance, compared with untreated Line 61 controls. Minami said plans to test AKST4290 in people with PD are in the works.
Researchers questioned Minami on AKST4290’s mechanism of action. Does it affect dopamine levels, which wane in PD? Minami said that while they are investigating whether the drug alters dopamine or its metabolites, she believes its benefit stems from anti-inflammatory properties. In support of this, Minami reported that AKST4290 markedly reduced microgliosis in the striatum of PD mice. In addition, the drug staved off the microglial activation and cognitive decline usually seen in response to chronic, low doses of lipopolysaccharide, a model of chronic inflammation.
Exactly how eotaxin in the blood manages to trigger neuroinflammation in the brain remains uncertain. Researchers in Tony Wyss-Coray’s lab at Stanford Medical School recently reported that the brain vasculature itself might play a role in transmitting such aging effects into the brain: expression of VCAM1, an adhesion molecule that tethers passing leukocytes, on brain endothelial cells was critical to allow aged blood to accelerate brain aging (Jul 2018 news).
Alkahest recently completed two Phase 2 clinical trials testing AKST4290 in people with age-related macular degeneration, another condition in which inflammation plays a starring role. Results from those trials are not yet public.
In addition, the company is testing GRF6019, a proprietary plasma fraction, in people with AD (see clinicaltrials.gov and clinicaltrials.gov), and has started a trial testing yet another fraction, GRF6021, in people with Parkinson’s disease with cognitive impairment or dementia (aka PDD).
At SfN, Alkahest scientists Eva Czirr and Viktoria Kheifets described how they identified the plasma fraction currently being tested in AD, GRF6019, as a booster of neuronal activity, synaptic function, and neurogenesis, and a douser of neuroinflammation, in mice and in neuronal culture models (Dec 2017 conference news and Jul 2018 conference news).—Jessica Shugart
No Available Comments
Part 1 of a two-part story. Click here for Part 2.
Tau comes in many forms, and calls many locations home, both inside and outside of cells. Which is the one that wreaks damage to neurons, and where exactly does it reside? Researchers grappled with these questions at back-to-back meetings in San Diego, the 5th RNA Metabolism in Neurological Disease Conference and the Society for Neuroscience annual meeting, on November 1–7.
Oligomers on the Move
One of tau’s infamous tricks is its ability to travel from neuron to neuron in the brain, by jumping the synaptic cleft (Feb 2012 news). First suggested by the characteristic spread of tangles in postmortem brain, and later backed up in mouse models, tau’s trans-synaptic spread has been considered a property of fibrils; however, in San Diego, Benjamin Wolozin of Boston University said tau oligomers travel in this way. He reiterated that oligomers are the most toxic species of tau.
Previously, Wolozin had reported that the RNA-binding protein TIA1 stabilized tau oligomers within stress granules, membraneless organelles that sequester mRNAs from translation during a stress response. Halving TIA1 expression reduced tau oligomers and their harmful effects in tau transgenic PS19 mice, even though these mice had even more tau fibrils than PS19 controls (Nov 2017 news). In San Diego, Wolozin added that TIA1 also facilitated propagation of tau oligomers throughout the brain.
In side-by-side preparations, Wolozin’s group generated tau fibrils and oligomers, then injected them into the entorhinal cortices of PS19 mice expressing either one or two copies of TIA1. Three months later, both oligomers and fibrils had seeded the spread of their kind to synaptically connected regions beyond the injection site. However, in the TIA1-deficient mice, the oligomers did not trigger spreading, whereas fibrils did, regardless of TIA1 copy number. Using antibodies specific for TIA1 and tau, Wolozin spotted TIA1 and oligomeric tau comingling in the neuronal cytoplasm, while fibrillar tau did not appear to interact with TIA1. Wolozin saw more neuronal loss in the entorhinal cortices of mice injected with oligomers than fibrils, suggesting that oligomeric tau is more toxic than fibrillar. The study was published on November 21 in Acta Neuropathologica (Jiang et al., 2018).
Wolozin proposed a bidirectional cascade, in which tau binding TIA1 promotes formation of stress granules in the cytoplasm, which shut down protein synthesis. In turn, the granules stabilize tau oligomers, furthering the cycle. Notably, Wolozin said that he observed oligomeric tau more commonly within the soma of neurons, while fibrillar tau accumulated in dendrites.
Oligomers, Take the Good with the Bad
Tau oligomers come in many shapes and sizes. Which are the worrisome ones? At SfN, Amritpal Mudher of the University of Southampton, U.K., described using biophysical techniques to track the emergence of different tau oligomer conformations throughout disease. Mudher uses flies to model tauopathy. These insects do not develop neurofibrillary tangles, but they do make tau oligomers, which is exactly what Mudher wants to study. Mudher reported that flies expressing three-repeat or four-repeat isoforms of human tau falter at climbing up glass vials and die younger than controls. The 3R isoforms are more toxic than the 4R (Sealey et al., 2017). Comparing the tau species present in fly neurons at early and late stages of this phenotype, Mudher found that oligomers emerge when symptoms first arise, while larger oligomers and ultimately fibrils appear only after the disease phenotype is entrenched. Mudher concluded that tau oligomers are primarily responsible for the deficits in the insects.
Are all tau oligomers equally harmful, or only certain strains? When Mudher treated the flies with lithium chloride, an agent that lessens tau phosphorylation and relieves damaging effects of tau pathology in flies (Mudher et al., 2004), she rescued the climbing and survival deficits, but found even more oligomers in the insects (Cowan et al., 2015). Reasoning that these oligomers must be of a distinct, and more benign, type than those found in sick flies, Mudher compared their conformation using Raman spectroscopy. By blasting molecules with a laser that jiggles molecular bonds, this technique produces a spectral signature specific to the secondary structure of a molecule. Mudher found tau oligomers in lithium chloride-treated flies to be distinct from those in untreated, diseased flies. Specifically, oligomers in the latter had a much higher β-sheet content than those in treated insects.
In collaboration with Martin Margittai of the University of Denver, Mudher also used Raman spectroscopy to compare several kinds of tau, including 3R, 4R, P301S, and delta-K18. Thus far she has only looked at fibrils, not oligomers, of these different types, but early findings already reveal striking differences, where each has a distinct combination of α-helical, β-sheet, and disordered content.
Mudher believes Raman spectrometry could identify the most toxic conformations of tau in disease models. That technique might even track tau oligomers, and disease progression, in diagnostic tests. Responding to audience questions, Mudher conceded that Raman requires grams of protein, so tau oligomers in CSF would need to be amplified to generate enough material for analysis. It is unclear whether the original oligomer conformations would be conserved after rounds of amplification.
Outside In, or Inside Out?
The field’s current emphasis on tau’s travels through the brain might easily obscure the fact that tau is an intracellular protein. Its normal post is on microtubules, and its abnormal accumulation in synapses correlates with synaptic dysfunction. How do extracellular tau oligomers relate to tau wreaking havoc within neurons? One possibility is that neurons take up these soluble forms of tau, which then travel to synapses. At SfN, Claudio Grassi of Università Cattolica del Sacro Cuore in Rome proposed another player. Grassi had previously reported that in cell culture, astrocytes more readily gobbled up tau oligomers than did neurons, and that tau accumulation in astrocytes interfered with release of astrocyte “gliotransmitters” that support synaptic function of nearby neurons. Grassi showed that tau accumulation in astrocytes blocked their release of ATP, which in turn stifled neuronal firing (Piacentini et al., 2017).
At SfN, Grassi described what happened when he blocked tau uptake into astrocytes with an antibody against GPC4, a heparin sulfate proteoglycan expressed only on this glial cell type. Such HSPGs have been implicated in the internalization of tau (Holmes et al., 2013; May 2018 news). Grassi reported that blocking astrocytic uptake of tau in hippocampal slice cultures prevented synaptic deficits otherwise caused by tau oligomers. He also reported that astrocytes can spit out tau, which is then readily internalized by neighboring astrocytes. He proposed that the indirect effects of astrocytic tau might explain a proportion of tau-dependent synaptic malfunction observed in neurons.
Eckhard Mandelkow of the German Center for Neurodegenerative Diseases (DZNE) in Bonn questioned Grassi after his talk, noting that the concentrations of tau oligomers Grassi used in his slice cultures far exceeded those found in the brain. He also challenged Grassi’s finding that the reduction in ATP released by tau-laden astrocytes was responsible for the synaptic defects in neighboring neurons. Previously, Eckhard and Eva Maria Mandelkow had reported that blocking the effects of adenosine, a breakdown product of ATP, with the adenosine A1 receptor antagonist rolofylline restored memory function in tau transgenic mice (Oct 2016 news). The Mandelkows had previously proposed that release of ATP by tau-laden neurons might increase extracellular levels of its breakdown product, which inhibits synaptic function in neurons. Grassi replied that he has not yet studied whether neuronal ATP or its breakdown products might contribute to his findings.
Mandelkow further pointed to a body of evidence implicating tau within presynaptic terminals of neurons (Apr 2015 news; Apr 2017 conference news). Researchers have recently reported that tau tethers to synaptic vesicles there, holding back their release (Feb 2018 news; Jul 2018 conference news). Mandelkow finds the hypothesis that tau harms synapses directly from within terminals more congruent with current data than the idea that it does so indirectly, via astrocytes. Grassi responded that he believes tau oligomers damage synapses via a combination of effects within astrocytes and the neurons themselves.
In synapses in the AD brain, tau is not the only troublemaker. Aggregated Aβ is commonly spotted at the scene, as well. For more on how synapses fare when both culprits are present, see Part 2 of this report.—Jessica Shugart
No Available Comments
Part 2 of a two-part story. Click here for Part 1.
Long before neurons die in the Alzheimer’s brain, their synapses go kaput. Both Aβ and tau pathology have been blamed for this synaptic breakdown, but at the Society for Neuroscience meeting, held November 3–7 in San Diego, researchers dissected how putting the two culprits together wields a blow quite distinct from either alone. They found that tau silenced neuronal activity, while Aβ stoked it. When placed together in the same mouse, tau’s silencing power won out. Curiously, tau turned down synaptic genes that operate specifically in excitatory neurons. And a new paper reported that a distinct gene-expression signature in excitatory neurons might explain why these cells are highly vulnerable to tau aggregation, as opposed to the relatively tau-resistant inhibitory neurons or glial cells.
At the Synapse, Do Aβ and Tau Clash or Conspire?
Both Aβ and tau aggregates have been blamed for synaptic dysfunction. What happens when the two meet? Two complementary lines of evidence presented at SfN suggest an answer more complex than simple synergy. Tau accumulation has been spotted in both pre-and postsynaptic compartments in autopsy brains, and also in mouse models of neurodegeneration. However, the field lacks models that illuminate the combined synaptic deficits evoked by tau and the other cardinal AD pathology, Aβ. To make one, researchers led by Marc Aurel Busche and Bradley Hyman of Massachusetts General Hospital in Boston crossed rTg21221 or rTg4510 mice, which express the wild-type or P301L mutated form of human tau, respectively, with APP/PS1 mice, which carry familial AD mutations in APP and PS1. They then asked the question, how do tau and Aβ interact to affect neuronal activity?
At SfN, Simon Dujardin first described analyses in the single strains. Measuring calcium transients related to spontaneous action potentials in cortical neurons, the researchers observed a distinct uptick in neuronal activity in APP/PS1 mice compared with wild-type controls. The opposite was true in rTg21221 mice, where neuronal firing was suppressed, and a large fraction of neurons appeared silent. Neurons were similarly shushed in rTg4510 mice, which develop neurofibrillary tangles owing to their expression of the aggregation-prone P301L mutant. These findings suggested that oligomeric forms of tau, as found in both the rTg21221 and rTg4510 strains, rather than NFTs, were responsible for the synaptic deficits observed in these mouse strains.

Loud Aβ, Quiet Tau. Neuronal activity was measured by the frequency of calcium transients in parietal cortices of control, APP/PS1, or rTg4510 mice. Neurons in APP/PS1 mice were hyperactive (red), while rTg4510 were silent (blue). [Courtesy of Busche et al., Nature Neuroscience, 2018.]
What would happen in the crosses? Dujardin reported that the neuronal hyperactivity in APP/PS1 mice was no match for the silencing power of tau. Regardless of whether the APP/PS1 were crossed to rTg21221 or to the tangle-bearing rTg4510 mice, neuronal firing in the crosses’ cortices was doused. Things got strange when the researchers turned off tau expression in six-month-old mice. After six weeks without transgenic human tau, the researchers were surprised to find that neurons were still hushed on the APP/PS1 background. However, in rTg21221 or rTg4510 mice, turning off tau was sufficient to return neuronal firing to normal. The researchers obtained similar results when they turned off tau earlier, in three- to four-month-old mice. They concluded that something about the presence of Aβ prevented the neurons from regaining their activity after tau was turned down. They hypothesized that when faced with both Aβ and tau, struggling synapses were less apt to recover.

Neurons in APP/PS1 animals crossed to either tau strain were relatively silent, compared with control or APP/PS1 mice (compare with previous figure). [Courtesy of Busche et al., Nature Neuroscience, 2018.]
The results appeared December 17 in Nature Neuroscience. The findings are based on overexpression models, but even so, Busche and colleagues write that therapeutic strategies aimed at either only Aβ or only tau might be confounded by the complex relationship between the two. Busche recently started a lab at University College London.
At SfN, Tara Spires-Jones of the University of Edinburgh, U.K., described her results from making similar crosses of tau and Aβ mice. Like Busche, Spires-Jones generated rTg21221 mice crossed to APP/PS1 animals. As opposed to a previous cross Spires-Jones reported, all the strains in her current study were generated on a tau knockout background to avoid confounds with endogenous mouse tau (Jackson et al., 2016). Spires-Jones compared AD pathology, gene expression, markers of synaptic loss, and behavioral deficits among the mouse strains. Compared with APP/PS1 mice, the APP/PS1 x rTg21221 offspring (a.k.a. MAPT-AD) had fewer, smaller plaques. Neither developed tau tangles, but the MAPT-AD mice had marked accumulation of tau in both pre- and postsynaptic compartments. Curiously, the MAPT-AD mice were hyperactive, running around more than control mice in open spaces.
Comparing the whole-brain transcriptomes of MAPT-AD, APP/PS1, rTg21221, and tau-knockout control strains, Spires-Jones reported a surge in expression of genes involved in inflammation, such as Trem2, Gfap, Cd68, and C1q, in both the APP/PS1 mice and MAPT-AD mice, over tau knockout controls or rTg21221 mice. Gene expression was no different between rTg21221 and controls. Though most of the pro-inflammatory gene-expression signature in MAPT-AD mice appeared in the APP/PS1 animals as well, a few inflammatory genes only revved up in the crosses, including Lilrb4, Ccl3, and Cst7.
The gene encoding cellular prion protein, PrPc, was strongly upregulated in the crosses. This drew the researchers’ attention because Aβ and PrPc reportedly interact at synapses, damaging them (Jul 2012 news). Strikingly, Spires-Jones also reported reduced expression of genes involved in synaptic function over each parental strain, suggesting that tau and Aβ pathology cooperatively turned them down. Spires-Jones speculated that this tau-induced decrease in transcripts encoding key synaptic genes could explain Busche’s data showing that tau dampens neuronal activity. When Spires-Jones switched off tau expression in 10-month-old MAPT-AD mice with doxycycline, these synaptic gene transcripts rose back to normal. Additionally, turning off tau relieved the animals of their hyperactivity.
However, turning off tau did not bring back synapses. Compared to tau knockout control or rTg21221 mice, both APP/PS1 mice and MAPT-AD mice had a third fewer synapses, especially near plaques, and they did not return after tau was switched off. Interestingly, Spires-Jones noted that rarely did Aβ and tau congregate in the same pre- or postsynaptic compartments. Aβ oligomers associated with synapses next to plaques, but tau accumulated equally in synapses near and far from plaques. The data is posted on bioRχiv.
“The impact of pathology on networks is of significant interest, and the use of models that have both amyloid and tau pathology, which better represent human AD, are a move in the right direction,” commented Karen Duff of Columbia University Medical Center, New York. “We still need to move toward more physiological levels of the proteins, with the correct spatial and temporal expression patterns to properly represent regional and cell-type vulnerability, but the interplay between the two pathologies is interesting,” she added.
In the manuscript, the scientists offer several explanations for synaptic loss orchestrated by Aβ and tau. It could be due to the inflammatory response, to association between Aβ and PrPc and, lastly, the downregulation of synaptic genes. Spires-Jones said that all downregulated synaptic genes—including AMPA and NMDA receptor subunits Gria2, Gria3, Gria4, Grin2a, Homer2, and Camk2b—function in excitatory, not inhibitory, neurons. This is in keeping with a recent study that reported a selective vulnerability of excitatory neurons to tau pathology (Jan 2017 news). Duff, Hongjun (Harry) Fu, and Abid Hussaini, also at Columbia, saw tau pathology accumulate in excitatory, not inhibitory, entorhinal cortex neurons of mice expressing P301L-tau. Excitatory cells selectively perished, dropping by as much as 70 percent in number compared with controls, while inhibitory neurons thrived.
A new study, led by Duff, Fu (now at Ohio State University), and Michele Vendruscolo at the University of Cambridge, U.K., provides a potential explanation for the excitatory neurons’ vulnerability. In the December 17 Nature Neuroscience, the scientists report that excitatory neurons in mouse and human brains express a cadre of genes known to coax tau into aggregation.

Tau’s Tangled Web. Groups of co-expressed genes associated with protein aggregation (red), tau tangles (green), aggregation protectors (blue) and promoters (yellow, upper left), tau (black), and genes shared between MS and tangles (brown). [Courtesy of Fu et al., Nature Neuroscience, 2018.]
The researchers extended the mouse findings linking tau accumulation to excitatory neurons to the human brain. They reported that in postmortem brain samples from AD patients compared with controls, tau pathology co-localized with markers of excitatory neurons, not inhibitory ones. This was true in the entorhinal cortex, where tau pathology starts, as well as in the surrounding neocortex, where tau spreads later in disease.
Why this selective vulnerability? To investigate, the researchers turned to two independent single-nucleus RNA-sequencing data sets—SNS and DroNc-Seq (Lake et al., 2016; Habib et al., 2017). These gene-expression data sets were generated by sequencing RNA from thousands of single nuclei. The nuclei were sorted from postmortem brain tissue of people who died without AD or any other neuropathology.
The researchers at Cambridge, led by Vendruscolo, found that nuclei hailing from excitatory neurons had a gene-expression signature distinctive from those derived from inhibitory neurons. In particular, the researchers identified opposing patterns of expression in a suite of previously identified genes making up a “protein homeostasis signature” (Aug 2016 news). Excitatory neurons had an overabundance of transcripts encoding proteins known to co-aggregate with tau or to promote tau aggregation. Conversely, they had a dearth of transcripts encoding genes that protect against tau aggregation. Nuclei from inhibitory neurons, or glial cells, had the opposite pattern.
In a co-expression network analysis, Fu and colleagues zeroed in on BAG3. This autophagy-related chaperone appears to orchestrate the aggregation protection network in inhibitory neurons (Behl, 2011). Working with Gail Johnson and Maoping Tang at the University of Rochester in New York, the researchers found elevated BAG3 protein levels in inhibitory compared with excitatory neurons in postmortem brain samples from people with and without AD. Further, modulating BAG3 expression in primary cultured neurons affected tau accumulation. BAG3 overexpression prevented tau accumulation, while knocking it down promoted a tau buildup. Duff said the researchers are now interested in why excitatory neurons are selectively vulnerable to tau homeostasis pathways and how that leads to tangle formation.
Spires-Jones commented that all three studies converge on the idea that pathological tau impairs excitatory neuron function. “The findings of Karen Duff and colleagues showing that excitatory neurons have a propensity to accumulate tau due to their expression patterns of genes involved in protein homeostasis are very interesting, and help explain why we observe accumulation of small tau aggregates in excitatory synapses both in our mouse model and in human AD brain,” she added. Spires-Jones also pointed out that while BAG3 levels were not changed in the MAPT-AD mice, transcripts encoding BAG5, a related protein, did drop.—Jessica Shugart
No Available Comments
The circadian clock—the body’s molecular timekeeper—is best known for tuning our sleep, metabolism, digestion, and body temperature to the cycles of dark and light. Yet it controls other functions, as well. At the Society for Neuroscience meeting held November 3–7 in San Diego, researchers reported that activation of astrocytes and microglia in the brain is one of those. Disrupting the clock led to untimely inflammation that destroyed synapses. Researchers proposed that this phenomenon could exacerbate harm done by Alzheimer’s pathology, and that correcting circadian imbalances might protect neurons in the face of neurodegeneration.
Previous studies led by Erik Musiek at Washington University in St. Louis have indicated that disruption of the circadian clock precedes cognitive symptoms in people with preclinical AD. In AD mice, knocking out the core clock protein, Bmal1, dampens the daily ebb and flow of Aβ in the hippocampus, hastening plaque formation (Feb 2018 news). Neuroinflammation is also known to wax and wane with the clock, as Musiek reported that knocking out Bmal1 throughout the mouse brain led to massive astrogliosis, neuroinflammation, and synaptic damage (Musiek et al., 2013). More recently, researchers got a similar, though less extreme, effect by deleting Bmal1 only in astrocytes. Not only did the cells become over-activated in the brain, they also no longer supported survival of stressed neurons in culture (Lananna et al., 2018).
At SfN, first author Brian Lananna presented unpublished findings extending this line of inquiry. He reported that in mice whose astrocytes express no Bmal1, astrocytes exhibited what has become known as an A1 astrocyte phenotype, revving up expression of a cocktail of pro-inflammatory genes. The researchers also noticed that expression of one gene, chitinase-3-like protein 1 (Chi3l1), was almost entirely snuffed out. This gene encodes YKL-40, a regulator of inflammation that is also an AD biomarker. YKL-40 levels rise in the plasma and CSF as disease progresses (Craig-Schapiro et al., 2010; Alzbiomarker YKL-40 (Plasma); Alzbiomarker YKL-40 (CSF)). In support of a link between Chi3l1 and the circadian clock, expression of the gene rose when the scientists knocked out two key regulators of Bmal1 expression, Per1 and Per2, in astrocytes. Chi3l1 expression rose during the day, while mice slept, and fell at night, while the nocturnal rodents scurried about their cages.
Lananna hypothesized that circadian control of Chi3l1 expression could explain why astrocytes become unhinged when their clock is disrupted. He generated Chi3l1 knockout mice to investigate. Those animals developed a similar astrogliosis phenotype as the Bmal1 knockouts. In response to the inflammatory instigator, lipopolysaccharide, astrocytes in Chi3l1 KO mice pumped out double the amount of pro-inflammatory cytokines, such as IL-6, as controls. Chi3l1 KO astrocytes also made poor neuronal nursemaids. When Lananna bathed cultured neurons in conditioned media from astrocytes, only media from normal astrocytes, not Chi3l1 KO astrocytes, bolstered neuron survival in the face of hydrogen peroxide stress. On the other hand, when the researchers grew neurons in media from astrocytes overexpressing Chi3l1, the cells gained extra protection.
Finally, Lananna asked whether Chi3l1 played a protective role in the brain of the APP/PS1 mouse model. When crossed to a Chi3l1 KO background, their hippocampi shrank more than did those of control APP/PS1 mice, despite similar Aβ burdens. Together, the findings suggested that astrocytic Chi3l1 dampens neuroinflammation and protects neurons from harmful stressors, including Aβ accumulation.
Following his talk, researchers asked Lananna more about Chi3l1 expression, and what explains increases in its protein product, YKL-40, with AD progression. He told the audience that according to the RNA-Seq analysis of the cerebral cortex carried out at the late Ben Barres’s lab at Stanford University, Chi3l1 was predominantly expressed in astrocytes, and not in other brain cells such as microglia or neurons. While little is known about its function in the brain, YKL-40 reportedly dampens inflammation in the lung by binding the IL-13 receptor (He et al., 2013). As to why it might increase as AD worsens, he suggested that, similar to anti-inflammatory cytokines like IL-10, YKL-40 might rise in step with the pro-inflammatory response to compensate for the damage the cytokines cause as disease progresses.
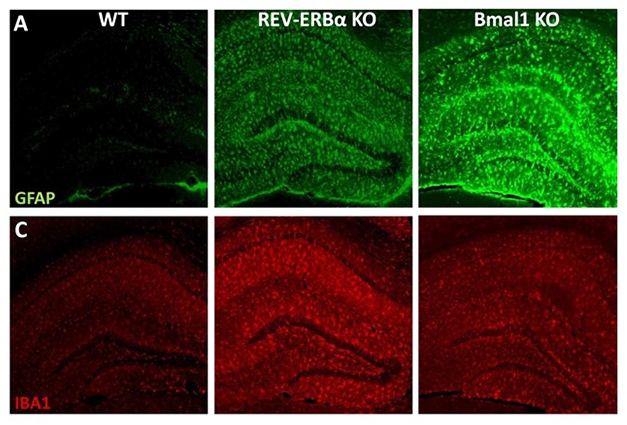
Glial Clocks Out of Sync. Knocking out REV-ERBα activates microglia (red, Iba1) and to a lesser extent, astrocytes (green, GFAP). Bmal1 KOs have more astrocytic than microglial activation. [Courtesy of Erik Musiek.]
While Bmal1 appears to promote Chi3l1 expression specifically in astrocytes, the master clock regulator also controls numerous other genes, many of which differ depending on cell type. To learn more about the pathways downstream of Bmal1, Musiek’s group also knocked out REV-ERBα. A nuclear orphan receptor, REV-ERBα is a key clock protein directly adjacent to Bmal1 in the circadian feedback loop. Bmal1 promotes expression of REV-ERBα, and in turn, REV-ERBα transcriptionally represses Bmal1. Knocking out Bmal1 reduces REV-ERBα expression by about 90 percent, while knocking down REV-ERBα elevates Bmal1 expression by only about 30 percent, Musiek told Alzforum.
At SfN, graduate student Percy Griffin reported that similar to Bmal1 knockout mice, the brains of mice lacking REV-ERBα were wracked with neuroinflammation and gliosis, and neurons in the hippocampus bore fewer synapses. However, the cause of the neuroinflammation appeared to arise from overactive microglia: expression of a host of microglial genes, including Iba1, TREM2, Ccl2, IL1b, and IL6, were elevated in the REV-ERBα knockouts. Complement proteins were also elevated.
Examining primary astrocytes and microglia in culture, Griffin found that knocking down REV-ERBα with siRNA did not alone spur astrocytes into activation. However, microglia did become activated by REV-ERBα knockdown, and when mixed with astrocytes, these microglia then instigated astrocytic activation as well. When the researchers collected media from these mixed glial cultures and added it to cultured neurons, they found that neurons grown in media from REV-ERBα knockdown glia were more sensitive to hydrogen peroxide stress than neurons grown in media from control glial cultures.
To home in on the specific role of REV-ERBα in microglia, Griffin is currently generating mice lacking REV-ERBα expression only in those cells. However, in the total REV-ERBα knockouts, Griffin already observed a strong disruption in circadian oscillations of microglial activation. In control mice, microglial expression of Iba1, a rough marker of activation, increases at night, while the animals are active. However, in the REV-ERBα knockouts, this rhythmicity is abolished, and microglia assume their nighttime levels of heightened activity 24-7. The researchers are currently looking at other activation markers and also assessing changes in microglial morphology throughout the day and night.
Also at SfN, researchers led by Rebecca Prosser at the University of Tennessee in Knoxville reported evidence that corroborated Griffin’s findings about the circadian oscillations of microglial activation, at least in the suprachiasmatic nucleus region of the brain, the home base of circadian rhythm research. There, similar to what Griffin saw in the hippocampus, microglia expressed more Iba1 at night. Examining microglial morphology, Prosser saw that, seemingly at odds with the finding that microglia expressed more Iba1 at night, they also stretched out their processes at that time, typically a sign of less activation. Prosser told Alzforum she was surprised by this, noting that interpreting morphological changes is not straightforward. Neurons in the suprachiasmatic nucleus are more active during the day—a rhythm that could influence microglia in this region.
Because microglia prune synapses—which can cause damage when in overdrive— Griffin and colleagues asked whether synapse numbers might also wax and wane in the mice, and whether that would change in REV-ERBα knockouts. Their preliminary findings, garnered from analyzing brain samples harvested at 10 a.m. (four hours after lights on) and 10 p.m. (four hours after lights out), indicate that synapse density is lowest when harvested at night. In REV-ERBα knockouts, this oscillation disappears, with synapses at a low level regardless of time of day, Griffin reported. The researchers have yet to connect this effect to oscillations in microglial activity or pruning. Still, they hypothesized that something about 24-7 microglial activation in REV-ERBα knockouts leads to synapse loss.
Could getting the clock back on track help stave off neuronal damage? The researchers proposed that as a nuclear receptor, REV-ERBα could make a therapeutic target. In fact, owing to its previously established roles in regulating metabolism in the liver, small-molecule agonists have already been developed. Griffin tested one of those, SR9009, in wild-type mice and in cultured cells (Solt et al., 2012). He reported that the drug dampened LPS-triggered neuroinflammation in the mouse hippocampus, and also promoted protection of stressed neurons in culture.
“The overall trend coming out of this research is that glial circadian clocks seem to regulate important processes in the brain, including neuroinflammation and synaptic pruning,” Musiek said, “If those go awry, that could potentially set the stage for cognitive impairment in the setting of disease pathology.”
While Musiek’s work suggests that circadian disruption could exacerbate damage caused by AD pathology, it is also possible that AD pathology messes with the clock. In support of this idea, researchers led by Inhee Mook-Jung at Seoul National University reported that in mice expressing human tau carrying the P301L mutation that causes frontotemporal dementia, tau pathology disrupted the oscillation of other key circadian functions, such as body temperature. The researchers tied this disruption to the degradation of Per2, a key clock protein that regulates Bmal1 expression.
Musiek told Alzforum that his lab is also investigating the relationship between tau pathology and circadian disruption. He added that just as the clock itself is controlled by tightly regulated molecular feedback loops, teasing out cause-and-effect relationships between disease pathology and circadian disruption will be a tall order.—Jessica Shugart
No Available Comments
No Available Further Reading
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.