CONFERENCE COVERAGE SERIES
Alzheimer's Association International Conference 2012
Vancouver, British Columbia, Canada
14 – 19 July 2012
CONFERENCE COVERAGE SERIES
Vancouver, British Columbia, Canada
14 – 19 July 2012
In Alzheimer’s disease drug development, how often does it happen that a large phase 2b trial of an experimental medicine cleanly meets both primary and secondary endpoints as set out in the protocol? Today, at the Alzheimer’s Association International Conference (AAIC) being held July 14-19 2012 in Vancouver, Canada, Dana Hilt of EnVivo Pharmaceuticals in Watertown, Massachusetts, reported that EVP-6124, a small-molecule partial agonist selective for the alpha-7 nicotinic acetylcholine receptor, improved cognition and function in a trial of 409 people with mild-to-moderate Alzheimer’s disease. The trial met seven of its nine endpoints with statistical and clinical significance, with trends on the remaining two. No posthoc statistical maneuvers were necessary to show that people who took the capsules once a day improved over baseline in a dose-dependent fashion.
While the field’s collective eyes are trained on immunotherapy, BACE inhibition, and early-stage trials studded with CSF or imaging markers, symptomatic treatments in mild to moderate AD are languishing in relative obscurity. “Treatments that target receptors in the cholinergic system in particular have been ignored,” commented Lon Schneider of the University of Southern California, Los Angeles. The results reported today suggest that, at least for cognition, the approach may work for patients who already take cholinesterase inhibitors and for those who don’t. The current trial hints, but does not formally show, that the improvement is clinically meaningful to patients; this issue remains to be addressed more extensively in a subsequent trial, Hilt said.
“I would be cautiously optimistic about this agent,” said Jeffrey Cummings of the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas, Nevada. “The trial met its prespecified outcomes and appears to be well conducted. Phase 2 results are not always confirmed in phase 3 but this agent deserves to be advanced.” Cummings is an adviser for EnVivo.
Previous trials in cognitively normal volunteers, people with schizophrenia and Alzheimer patients had suggested that EVP-6124 enhances cognition and acts at lower doses than other compounds targeting the same receptor. For example, an agonist called RG3874 by Roche, which was discontinued in February 2011, was given at doses ranging from 5 to 50 milligrams per day (Mancuso et al., 2011). The trial reported today at AAIC was a dose-ranging study of 0.3, 1, and 2 mg once a day. Empirically, toxicity from pharmaceuticals is rare when given at doses well below 10 mg per day.
The trial sought patients in their 70s whose MMSE fell between 14 and 24 in hopes of detecting a treatment effect at the point in disease progression where patients tend to worsen fastest. Steep decline means a drug effect is easier to measure with standard cognitive batteries, which are less sensitive in very mild than in moderate AD. At baseline, however, the patients ranged quite widely, from a starting MMSE of 11 to a high of 29, meaning the trial did include some very mild patients.
The trial used the ADAS-cog 13 as primary endpoint; the most important secondary endpoint was the CDR-SB as a functional measure. Other secondary endpoints included the ADAS-cog 11, other cognitive tests, the NPI, and the ADCS-ADL as a global measure. In addition, the trial protocol pre-specified analysis with three composite measures developed by John Harrison, a neurospychologist and statistician at Metis Cognition in Kilmington, U.K. These were meant to probe whether the drug exerts specific effects on cognition, memory, or executive function that the broad ADAS-cog collection of tests as a whole might obscure, particularly in earlier-stage patients.
Click image for larger view
Conducted at 30 sites in the U.S., Russia, Romania, Ukraine, and Serbia, the trial consisted of 60 percent of patients in the U.S. and 40 percent in Eastern Europe. Results were nearly identical between the two continents, Hilt said. The trial used no biomarkers other than plasma EVP-6124 to check whether patients actually took the prescribed pills. Patients were allowed to join regardless of whether they smoked (a habit that affects nicotinic receptors; 8 percent did) or whether they took cholinesterase inhibitors, which do not target the α7 receptor directly but nonetheless influence the cholinergic system. About half of the patients in this Phase 2b trial added EVP-6124 to existing therapy, while the other half took the agonist alone. “We meant for this study to reflect a neurologist’s ‘inbox,’ i.e. include a real-world sample of the kind of patients a neurologist or other clinician would see,” Hilt said.
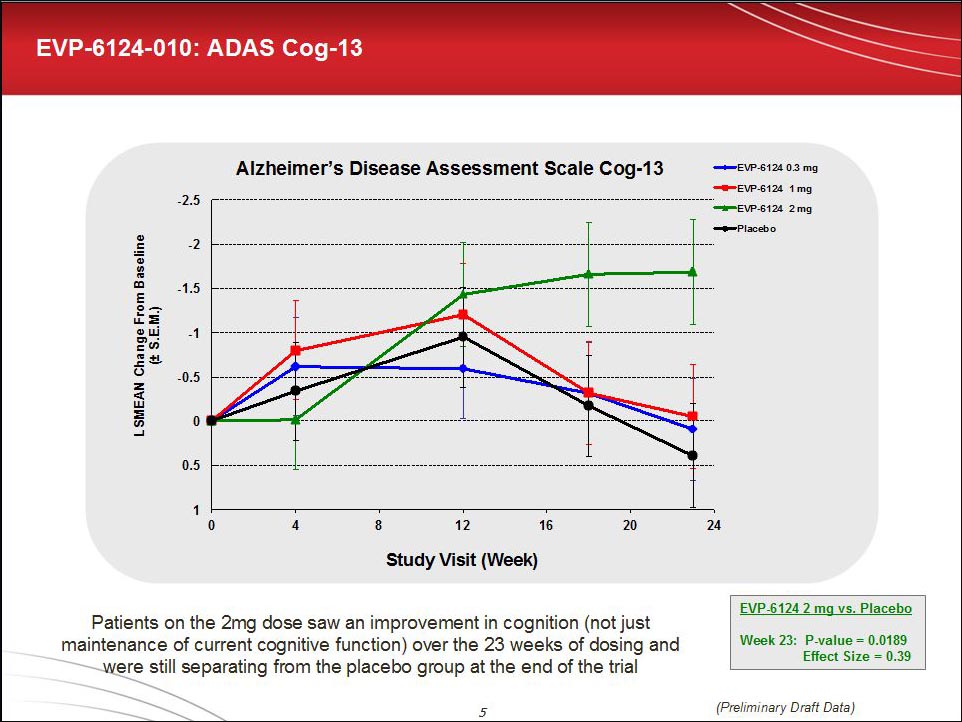
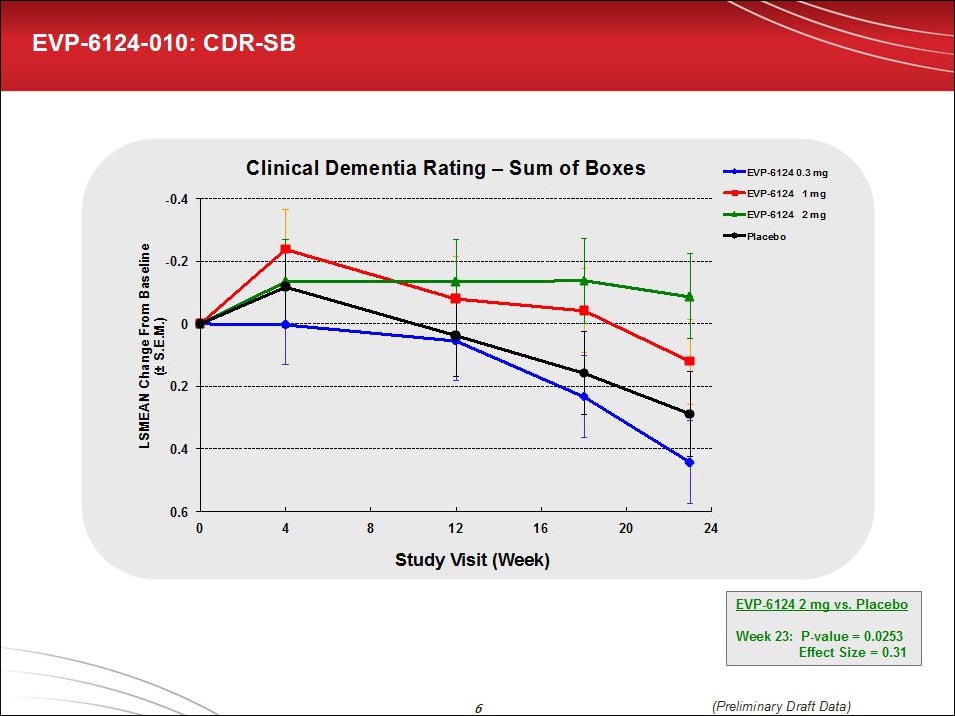
In Vancouver, Hilt reported that all primary endpoints except for the ADCS-ADL and the MMSE reached statistical and clinical significance in both de novo and add-on patients. This analysis was done in the intent-to-treat population, not only in those who completed treatment. The effect size for the ADAS-cog-13 was 0.39 with a p value of 0.0189, Hilt reported. For the Clinical Dementia Rating-Sum of Boxes, those numbers were 0.341 and 0.0253, respectively. (By comparison, the effect size for cholinesterase inhibitors range between 0.15 and 0.28 depending on the dose.)
The EVP-6124 numbers improved when the scientists analyzed the data with composite endpoints developed to measure specific treatment effects in patients with milder disease, who tend to show little overall decline in 6-months and hence barely move the needle on the ADAS-cog. For the 2 mg group, the cognition composite score was statistically significant at 0.0037, the memory composite at 0.0088 and the executive function composite at 0.0427. Of these three domains, the effect size was highest for cognition at 0.42, confirming again that the drug is primarily a pro-cognitive agent, said Hilt. These analyses, too, were pre-specified in the study’s statistical analysis plan.
The time course of participants’ improvement drew attention at the conference. On the primary outcome, all groups first improved from baseline, probably due to the placebo effect and optimism about being in a therapy trial. All groups stayed together up until 12 weeks; after that, the curves separated. By 23 weeks, the last time point measured, only the 2 mg dose stayed up and apart from placebo. “This is a cautionary finding for Phase 2 studies to run beyond 12 weeks,” Gerhard Koenig of EnVivo told Alzforum.
Importantly, Hilt said, the results showed a graded response with different doses, a feature drug developers want to see. The 0.3 dose was ineffective, the 1 mg dose reached significance on some measures, and the 2 mg dose was most efficacious.
For historical perspective, scientists pointed out that the pivotal trial for approval for donepezil derived its effect size to a larger part than EVP-6124 from a decline in the placebo group, with the treatment group improving approximately by 0.5 points over baseline. In contrast, in the EVP-6124 trial the placebo group hardly declined over 6 months, which has become typical as milder patients are increasingly entering trials. Instead the drug-placebo difference arose from 1.9 point improvement of the 2 mg group over baseline.
The clinical dementia rating sum of boxes, a functional measure, showed a dose effect. By comparison, the cholinesterase inhibitor donepezil had no effect on this measure in its pivotal trial. The CDR-SOB measure is increasingly widely used in clinical trials, though it is not established yet as an outcome the FDA will accept as a basis for approval. For that purpose, the ADCS-ADL is a more accepted endpoint. It trended in the right direction with a 0.2 effect but missed statistical significance in this trial.
Click image for larger view
On safety, Hilt reported that the drug is safe and well tolerated. Mild nausea and constipation did occur in all groups and more so in the treatment groups. Analysis of the adverse effects in the various groups – people on acetylcholinesterase inhibitors, people on EVP-6124, people on both drugs, aged controls on placebo – suggested that taking a cholinesterase inhibitor alone cause more gastrointestinal trouble than taking the alpha 7 agonist. Adding the agonist to donepezil or rivastigmine did increase the incidence of these side effects. Four percent of placebo patients and 7 percent of 2 mg EVP-6124 patients reported these side effects. Very few of these led to dropout and most resolved on their own, Hilt said. In essence, the scientists claimed, EVP-6124 appears more tolerable than current therapy. Previous trials in a younger population with schizophrenia caused no adverse events at all, Koenig noted. Eighty percent of participants completed the trial, and there was no difference in the fraction completing in the different treatment groups.
The next challenge lies in demonstrating what the cognitive improvement means for the patient, Koenig said. “This drug safely improves cognition. This much I can say, as it’s been shown consistently in all trials we conducted. But we need to do more work to be able to show formally how this matters for the patient.” A longer Phase 3 trial perhaps testing a higher dose will start in 2013. It will experiment with patient-reported outcomes and additional measures to address whether this drug is clinically meaningful for patients. EVP-6124 sensitizes the receptor to acetylcholine rather than stimulating it directly. For details on the drug’s mechanism of action, see prior prior ARF news story.—Gabrielle Strobel.
The going hypothesis on amyloid-β forms states that oligomers are the most toxic species, and yet no one can robustly measure them in human CSF, where oligomers are rare clumps floating in a sea of monomer abundance. If researchers could quantify the oligomer population in a person, then they might get a solid clue as to the person’s Alzheimer’s or cognitive impairment status, as well as a measure of whether anti-amyloid therapies are doing any good. At the Alzheimer’s Association International Conference, held 14-19 July 2012 in Vancouver, Canada, two companies presented their progress toward oligomer-measuring assays. Merck’s West Point branch in Pennsylvania and the biotech company Amorfix, of Mississauga, Ontario, are taking distinct approaches to pick up femtogram amounts of oligomers. Both are observing similar trends in human cerebrospinal fluid samples. The assays are in early stages, and the designers are planning more work to validate them.
The field has been chasing the Aβ oligomer concensus for several years. In fact, a consensus on oligomers has been so elusive that it has been derided as the emperor without clothes (Benilova et al., 2012). One approach is to perform an enzyme-linked immunosorbent assay (ELISA) using the same Aβ antibody as both the capture and detection reagent. If the antibody can only bind a monomer at one site, then any conglomerate detected in this manner must be a dimer or more (see ARF related news story on Xia et al., 2009). For example, IBL International, based in Hamburg, Germany, sells such a single-antibody kit. However, this approach is not very sensitive, commented Kevin Felsenstein of the University of Florida, Gainesville, who was not involved in the current AAIC presentations but has collaborated with Amorfix in the past. IBL’s kit detects oligomers in the picomolar range, but CSF samples—particularly those from healthy people—may contain far less than that. With CSF containing 1,000-fold more monomer than oligomer, noted William Goure of Acumen Pharmaceuticals in Livermore, California, oligomer detection must be exquisitely sensitive and selective.
Merck’s approach, presented on a poster by Mary Savage, is to apply an ELISA-like technology using an antibody specific for oligomeric Aβ. The company’s 19.3 antibody, developed in collaboration with Acumen, preferably binds to synthetic oligomers over monomers. It was originally made in mice, but the researchers have modified its sequence to make it more human-like. Its epitope is unknown, but Savage suspects it binds to a three-dimensional conformation only present in oligomers. No other humanized amyloid antibody bests 19.3 for oligomer selectivity, Goure said.
Savage and colleagues first tried using 19.3 to capture oligomers in a typical ELISA platform, with a pan-amyloid antibody called 82E1 as the detecting antibody. That worked, but could only pick up oligomers reliably at 4.2 picograms/milliliter or more—fine for brain tissue but not good enough for CSF samples. The researchers turned to a more sensitive assay developed by Singulex of Alameda, California. Called Erenna, this ELISA sandwiches the antigen and antibodies between a magnetic bead and a fluorescent tag, then quantifies the fluorescence pulled down by the magnetic beads. With this ELISA, the team was confident they could detect as little as 420 femtograms of oligomers per milliliter of CSF. The assay’s sensitivity exceeds that of previously reported CSF oligomer assays, the scientists said.
In preliminary tests with a handful of purchased CSF samples, Savage was excited to see that people with AD had more oligomers than did young control donors. A further 72 samples confirmed the assay was able distinguish the two populations, with less overlap between the groups than the field is used to seeing with CSF monomer ELISAs. “You can start to draw a line” differentiating the populations, Savage told Alzforum. “I think this might have some utility for diagnosis.” In collaboration with groups in Norway and in the U.K., Savage will next test the assay in longitudinal cohorts that also collect other fluid, imaging, and cognitive markers.
However, there is still fine-tuning to be done. Many control samples had fewer oligomers than the researchers could confidently measure. Savage hopes to improve the ELISA by treating the CSF samples with a dash of detergent; without it, oligomers stick to the walls of the tubes in which they are stored (Pica-Mendez et al., 2010). In addition, the oligomer standards she used likely contained some monomer, so purer standards might improve the assay, she said (Hepler et al., 2006).
The assay is not currently available to researchers outside Merck. Merck does not typically produce diagnostics, and this assay was originally intended as an internal companion biomarker for a therapeutic antibody development program that has since been discontinued. Savage said that if further validation proves the assay’s worth, she would like to find a way for others to use it.
Amorfix took a different tack, measuring oligomers indirectly, as presented in a July 16 talk by Marni Uger. They have already commercialized a size-filtering method to quantify mouse amyloid oligomers, which is available from JSW Life Sciences. To this technique, they added a monomer-masking method developed in the laboratory of Amorfix founder Neil Cashman of the University of British Columbia in Vancouver. The researchers treat CSF samples with a molecule that modifies epitopes exposed in monomers, preventing detection by their amyloid antibody. The idea is that only epitopes buried inside of oligomers escape the modification, while monomers are tagged and become invisible to the antibody. Then, the researchers dissociate the oligomers and measure unmodified monomer concentration with an ELISA method that, like Merck’s, measures the fluorescence signal from antigens pulled down by magnetic beads.
Amorfix claims it can detect as little as 44 femtograms of oligomeric amyloid-β. In nearly 200 purchased CSF samples, the assay distinguished people with mild cognitive impairment (MCI) from those with AD, and each disease state from age-matched healthy control samples. MCI samples reportedly had the highest oligomer signal, with AD falling between MCI and control levels. “The strength of our assay is the ability to distinguish the MCI individuals,” Uger said, since those are the people who might benefit most from early treatment. Cashman and Uger speculate that oligomers might be most prevalent in the early stage of neurodegeneration because they are produced by neurons under attack from the disease.
However, biphasic assays can be difficult to interpret. For example, a mid-range oligomer value alone might indicate a person is either in the early stages of MCI or on the way to AD dementia. Clinicians would have to use the oligomer measurements in conjunction with clinical symptoms to assign the right stage of disease, Cashman said.
Taken together, preliminary data from the two teams indicate that people with AD had approximately two to five times the amount of CSF oligomer as controls; this stands in contrast with monomer Aβ, which falls as one approaches AD. However, the new techniques require extensive validation. Meanwhile, Felsenstein sounded a note of caution about oligomer measurements. “It may be artifact,” he said. One problem is that storing CSF might alter the proportion of monomers and oligomers; another is that detergents and salts added in processing can skew oligomer counts. The best approach, Felsenstein suggested, is to assay fresh CSF and limit manipulation to simple dilution of the sample.—Amber Dance.
No Available Comments
At the Alzheimer’s Association International Conference held 14-19 July 2012 in Vancouver, Canada, Gabrielle Strobel met up with Luca Santarelli, Senior Vice President and Head of Neuroscience at Hoffmann-La Roche. Santarelli is perhaps best known for his work on depression—while a young scientist at Columbia University, he discovered that antidepressants such as Prozac work by inducing neurogenesis (Santarelli et al., 2003). But Santarelli moved into drug discovery research at Roche/Genentech in 2005 and now oversees the Alzheimer’s therapy development program, among others, at the parent company in Basel, Switzerland. Below are excerpts from the conversation, updated after news on the first bapineuzumab trial broke on 23 July.
Q: There is concern in the Alzheimer’s disease research community about pharma getting out of CNS, particularly AD. What is Roche’s position?
A: We are very much into this area. We have a large CNS program. For example, we have gantenerumab in Phase 3. It is the first monoclonal antibody targeting prodromal AD. The study’s name is SCarlet RoAD. It is in the middle of recruiting 770 patients and will complete recruitment next year. [Editor’s note: See on ClinicalTrials.gov.] The strategy is to go early in the course of disease where we hope to achieve most benefit.
Q: What else is different about it?
A: A significant percentage of patients involved in AD studies, even at the mild to moderate stage, do not display amyloid pathology. This diagnostic inaccuracy almost certainly adds noise to these trials. In SCarlet RoAD we are identifying prodromal AD patients by deploying a molecular diagnostic strategy. In this case, we use CSF Aβ along with cognitive testing to identify the patients with underlying AD pathology. For the current trial, we use a commercially available research-grade test, but we are developing a companion diagnostic assay test for future use.
Q: What makes you confident that gantenerumab will work?
A: This compound has published results showing that it reduces brain amyloid three times faster than other published amyloid-lowering agents. [Editor’s note: See Ostrowitzki et al., 2011.] The antibody binds to aggregated Aβ in fibrils and plaques. It is fully human, and the trial uses a subcutaneous formulation. If positive, the study could be pivotal.
Q: Is this Phase 2/3 going to be enough for a New Drug Application?
A: Confirmation in a separate trial is often required, but that depends on the effect size of your pivotal trial, and on the perceived medical need and other regulatory considerations at the time the data become available. We are now assessing what additional trial will be appropriate.
Q: How are the talks with the FDA going?
A: In our interaction with the FDA, we have experienced a collaborative spirit, where the agency is open to new ideas and willing to give us helpful advice on how to make progress on these novel trial designs. For example, the SCarlet RoAD trial does not use ADAS-cog. We use CDR-SOB; it is a single primary that is a global measure with some functional components.
Q: To read the trade and general media, you’d think the entire world turns on the results of the bapineuzumab and solanezumab Phase 3 trials that will read out in the next few months. Indeed, the first of four bapineuzumab trials on 23 July reported negative topline results in ApoE4 carriers (see ARF related news story). There’s considerable worry for the future of anti-amyloid drug development if these Phase 3 trials show no clinical significance. What do you think can be reasonably expected from them?
A: It is hard to predict the outcome of the additional Phase 3 trials. We will have to be patient and wait for the results. Hopefully, the outcome of the remaining trials will help provide support to the amyloid hypothesis. I would add, however, that in AD it is important to use doses that have previously demonstrated amyloid removal in patients; patients should be included early in their illness, at the prodromal stage or even earlier; and biomarkers indicative of Aβ amyloid pathology should be used to confirm the diagnosis before randomization.
Q: Venture out and make a prediction, will you?
A: I see three scenarios. 1) Bapineuzumab meets the endpoint in the ApoE4 non-carriers and the trials form basis for approval. That seems optimistic. 2) We get a mixed result with a signal but no basis for approval. That seems more likely. 3) The program is totally negative. The first two are good news for the field. The last would be a setback. But there are other approaches, like going earlier, that might yield good results and that are getting underway. The field will have to wait longer to gain confidence. Incidentally, the intense attention focused on bapineuzumab reflects also some of the challenges of charting a new path.
Q: Will Roche’s approach and investment into gantenerumab change if bapineuzumab and/or solanezumab were to be negative?
A: We will continue. As far as gantenerumab, we have targeted a different population, and there is a fundamental issue when targeting this disease late. [Editor’s note: Roche’s internal communication rules prevent Santarelli from commenting on Genentech’s crenezumab, which is being tested in patients with mild to moderate AD; see the ABBY Trial. See also ARF related news story.]
Q: The gantenerumab trial is viewed as innovative because it enrolled a prodromal population based on memory impairment short of dementia and a CSF Aβ cutoff. How much confidence do you have in the CSF biomarker assays that are currently available? There is discussion about their center-to-center and lot-to-lot variability, and none of them are FDA validated.
A: I am very confident in them. Just think about the mild to moderate trials. These studies are using clinical criteria to include patients, which means an estimated 20 to 30 percent of patients randomized in these trials do not have amyloid pathology. That was true—some 20 percent amyloid-negative people in the AD group—even in ADNI. Even with their limitations, the current assays do increase the homogeneity of your group considerably. You have 90 percent of people who truly have prodromal AD because you filter the clinical diagnosis through a biomarker. The ADNI and work from multiple longitudinal studies worldwide have really shown that. Some people think that accuracy of diagnosis may be even more important to show treatment benefit than early stage.
Q: There is great emphasis in the field on standardizing biomarkers to where they are robust and stable for use in multicenter trials and clinical care. The FDA issued a Guidance on Companion Diagnostics last year, but the concept is not broadly known among the wider research community. Roche being in part a diagnostics company, how do you approach biomarker development for your AD portfolio?
A: For us it is important to identify a way to diagnose AD early that is reproducible, high quality, and can become a gold standard for hospitals everywhere in world. That is how we approach this. Being both a pharma and a diagnostic company, we have a unique opportunity of developing a diagnostic and using it to evaluate the efficacy of our compound. Our strategy for AD is now focused on CSF, but we are also looking very actively in plasma.
Q: Have you found anything in plasma yet?
A: Nothing definitive yet. We are working with a number of companies.
Q: Besides gantenerumab and crenezumab, what else does Roche have in its pipeline for neurodegeneration?
A: We have an oral BACE1 inhibitor in Phase 1. There, we are in a pack with others. We also have a monoamine oxidase B (MAO-B) inhibitor in a Phase 2b dose-ranging study. This is a compound that targets the inflammatory aspect of age-related neurodegeneration. We have internal data showing that this mechanism is beneficial for AD. The inhibitor’s properties are linked to reducing the release of reactive oxygen species. This compound was discovered by Roche, then taken over by Evotec, which conducted trials for a different indication, and more recently, we took it back to evaluate it in AD.
Q: How about preclinical?
A: In the preclinical stage, we are active on several different fronts on AD. For example, we have two monoclonal antibodies that target tau, one internally developed at Roche, and one discovered at AC Immune. Additionally, we are developing a series of molecules that are highly brain penetrant. We use a technique we call brain shuttle. [Editor’s note: See ARF related news story.] The tau antibody is one molecule we are developing that way. Tau is thought to be released from cells and from there to spread to other cells, so you will need a technology that allows you to get a high amount of therapeutic antibody in, more than the 0.5 percent or so that we have seen so far. Brain shuttle has demonstrated the potential to greatly increase brain antibody concentration.
Q: What about combination trials? Researchers increasingly say it may be too much to ask of any single treatment to dramatically improve symptomatic AD. The FDA issued a guidance already in 2010 calling for trials of two unapproved drugs simultaneously in serious diseases including cancer and AD. You could combine an amyloid antibody and a BACE1 inhibitor, an antibody and your MAO-B inhibitor, or an amyloid and a tau antibody. Pipedream? In the works? Is Roche interested?
A: We were just discussing whether to do this for gantenerumab and our BACE inhibitor, in fact. There you’d have synergy between clearance and production. It should be possible. The problem with this example is that there is a time delay, so we may have to gather more data on the BACE inhibitor before we take that tack. We have that approach in oncology. For AD, it’s not out of the question at all.
No Available Comments
Think Russian nested dolls. Researchers at the University of Manchester have discovered a mutation within the mutation that is the C9ORF72 expansion causing amyotrophic lateral sclerosis (ALS) and frontotemporal dementia. At the Alzheimer’s Association International Conference, held 14-19 July 2012 in Vancouver, Canada, Stuart Pickering-Brown of the University of Manchester presented two cases with an apparent misspelling in the hexanucleotide repeat expansion. Two brothers, both of whom had frontotemporal lobar dementia (FTLD), carried 800 or so repeats in C9ORF72, but the repeat’s usual GGGGCC sequence does not seem to be present. The researchers have not yet determined the sequence in this case, but suspect the last two cytosines are altered.
Pickering-Brown, speaking on 15 July, described an investigation into the genotypes of nearly 400 people with FTLD. The normal number of repeats in C9ORF72 is 23 or fewer; 8 percent of the Manchester cohort possessed larger stretches of up to 1,500 repeats. The C9ORF72 expansions cause a specific pathology consisting of TDP-43-positive inclusions in the brain or spinal cord, as well as ubiquitin-positive but TDP-43-negative aggregates in the cerebellum.
The mystery of the two brothers arose when pathologist David Mann examined their autopsy samples and did see the telltale cerebellar pathology, but, curiously, the genetic analysis showed only half a dozen repeats in C9ORF72. Pickering-Brown recalled that Mann queried, “Are you sure these do not have a repeat expansion?” By repeat-primed polymerase chain reaction, the method of choice to identify the expansion in this difficult-to-sequence location, they were not. The PCR output showed six peaks, corresponding to six repeats. But when the researchers scrutinized their results more closely, they observed “really tiny peaks” that could indicate longer expansions, Pickering-Brown told Alzforum. Southern blots confirmed Mann’s hunch: The C9ORF72 gene was, in fact, 800-950 repeats long in the brothers. “Something is stopping the repeat-primed PCR from working efficiently,” Pickering-Brown concluded.
A mutation in one of the sequences to which the PCR primers bind could interfere with the subsequent chain reaction, he reasoned. One primer binds outside the repeat region, but the sequence there was normal. The team surmised that the mutation must be present inside the hexanucleotide repeats themselves. They designed primers with nucleotide substitutions at each of the six sites in the GGGGCC code. When they altered each of the last two cytosines individually, the PCR worked “a little bit better,” but still not as well as it ought to. Although the team is not sure, they suspect that both of those final bases are altered in the DNA from the two brothers.
“Stuart’s data demonstrated that extensive Southern blot analysis is going to be required to answer key issues related to C9ORF72,” commented Leonard Petrucelli of the Mayo Clinic in Jacksonville, Florida, in an e-mail to ARF. “Variation in the repeat sequence in C9ORF72 suggests that this cause of FTD and ALS might be more common than we first thought,” added Petrucelli, who was not involved in the study. For his part, Pickering-Brown thinks this altered repeat sequence is quite rare, although he will not be able to confirm his hunch until he knows the right code to look for in other samples. How did one family acquire the unusual repeats? Perhaps, Pickering-Brown speculated in an interview with Alzforum, the polymorphism arose in this particular kindred even before the repeat expansion occurred.
People who carry C9ORF72 expansions share a common haplotype, leading many researchers to suspect that the repeats proliferated in a single founder (Smith et al., 2012). Pickering-Brown’s work, albeit incomplete, could support an alternative theory. It might be that something about this haplotype promotes the hexanucleotide expansion, which would explain how the repeats expanded separately in the family he studied. Corroborating this idea, Pickering-Brown and others have found that the length of affected people’s repeat regions varies widely among their tissues. He presented Southern blots of brain homogenates, which show that the C9ORF72 gene appears not as a single-sized band but a faded smear ranging from several hundred to a few thousand repeats long. Thus, the repeat region may be inherently unstable and prone to shrink or expand with every cell division. As Petrucelli concluded, “The elusive chromosome 9 locus is still not giving up its secrets easily.”—Amber Dance.
No Available Comments
Many had trouble reaching the brain. Or they got in but couldn’t stay, ousted in short order by P-glycoprotein. At long last, drug developers have overcome these and other hurdles, and well over a decade of effort developing β-secretase (BACE1) inhibitors is starting to pay off. At the Alzheimer’s Association International Conference held 14-19 July 2012 in Vancouver, Canada, three companies reported promising Phase 1 data on brain-penetrant compounds that block BACE1 activity. The protease cuts amyloid precursor protein (APP) to kick off production of the Aβ peptides believed to be a molecular culprit of Alzheimer’s disease. “Even though it’s been very difficult to develop BACE inhibitor drugs, they are now coming along. They are in clinical trials. They can get into the brain, and they inhibit BACE1 and reduce Aβ levels dramatically in the CSF. I’m very excited about the possibilities,” said Robert Vassar of Northwestern University Medical School in Chicago, whose lab cloned and characterized BACE1 (Vassar et al., 1999).
Crystal structure of BACE1, a leading target in AD drug development research. Image courtesy of Wikimedia
Eli Lilly and Co., Indianapolis, Indiana, appeared to set the pace for the newest batch of BACE1 inhibitors. At last year’s AD/PD International Conference in Barcelona, Spain, and later in a Journal of Neuroscience paper, the company reported that it finally had in hand an oral compound (LY2811376) with nice drug properties that got into the brain and reduced Aβ in healthy volunteers (see ARF related conference story and May et al., 2011). Unfortunately, that molecule never reached Phase 2. Rat studies linked the Lilly compound with retinal pigment epithelial defects—perhaps the only consolation being the absence of these problems in BACE knockout mice, which indicated they were an off-target effect and that blocking BACE1 could still conceivably work as an Alzheimer’s intervention.
In Vancouver, Lilly scientists reported preclinical and Phase 1 data on a new BACE1 inhibitor, as did Eisai and Merck. In addition, though not presenting at AAIC, Roche has an oral BACE1 inhibitor (RG7129) in Phase 1 (see Santarelli Q&A), and High Point Pharmaceuticals, North Carolina, is recruiting people with mild cognitive impairment (MCI) or mild AD for a 28-day course of its BACE1 inhibitor (HPP854) in a Phase 1 safety study. The Philadelphia-based biotech company Vitae Pharmaceuticals is partnering with Boehringer Ingelheim to develop oral BACE compounds that lower brain Aβ in AD mouse models. Other companies including AstraZeneca, CoMentis, and Takeda Pharmaceutical Company are also developing BACE inhibitors (see ARF related news story), but did not present at AAIC this year. Genentech has an entirely different approach—bispecific antibodies with one arm targeting BACE and the other recognizing transferrin receptor to boost brain penetrance (see ARF related news story on Atwal et al., 2011, and Yu et al., 2011). Below is a summary of the data Lilly, Eisai, and Merck presented at AAIC.
Lilly’s Patrick May reported that the compound LY2886721 showed good selectivity (i.e., did not inhibit other aspartyl proteases such as cathepsin D, pepsin, and renin) and reduced Aβ in a dose-dependent manner in HEK293Swe cells and in primary neurons from PDAPP mice. Despite a short half-life of three hours in mice, a 3-30 mg/kg dose lowered brain Aβ by 20 to 65 percent, relative to vehicle-treated groups, and the effect lasted up to nine hours after dosing. In beagle dogs, where the molecule has a longer half-life, a 0.5 mg/kg dose halved cerebrospinal fluid (CSF) Aβ in nine hours, and plasma Aβ levels were still down after 24 hours. Before moving into CSF biomarker studies, the Lilly team conducted an initial single ascending-dose study evaluating safety, tolerability, and of plasma Aβ pharmacodynamics following 1-35 mg oral doses of LY2886721 to healthy adults.
In Phase 1 studies, people took the compound by mouth, then had CSF samples collected at baseline and every one to four hours for 36 hours (single-dose study), or 24 hours after the final dose (multiple-dose study). Scientists measured CSF levels of APP’s non-amyloidogenic (sAPPα, C83) and amyloidogenic (sAPPβ, C99) cleavage products. The single-dose trial enrolled healthy adults for LY2886721 (10 or 35 mg), or placebo, and participants in the multiple-dose study got 5, 15, or 35 mg of the inhibitor, or placebo, once daily for two weeks. The compound lowered CSF Aβ40, Aβ42, and sAPPβ concentrations, while increasing sAPPα, consistent with what is expected for BACE1 inhibition, Lilly’s Brian Willis reported. On a poster by Ferenc Martenyi and colleagues, the inhibitor showed good pharmacodynamic (PD) and pharmacokinetic (PK) properties, and was safe and well tolerated up to six weeks from final dosing in the 68 subjects who completed the Phase 1 studies. The compound has entered a six-month Phase 2 trial of the two higher doses in early AD patients whose disease is ascertained with a positive brain amyloid PET scan by florbetapir, May said in his talk.
Eisai is also developing an oral BACE inhibitor (E2609), and the main news so far is much the same. According to company scientists, it is highly potent and selective, crosses the blood-brain barrier, and seems safe and well tolerated in healthy people thus far. It did well enough in early clinical studies to progress to Phase 2. Eisai’s single-dose Phase 1 trial assessed safety and plasma readouts only; its multiple ascending-dose trial included CSF measures as well. In the single-dose study, young healthy volunteers received 5, 10, 25, 50, 100, 200, 400, or 800 mg of inhibitor, or placebo, while an elderly cohort got a single 50 mg dose. The scientists collected blood samples before dosing and at various times up to 144 hours afterward. Plasma Aβ1-x levels dropped in a dose-dependent manner, with 52 percent clearance for 5 mg doses and 92 percent at 800 mg. At higher doses (200-800 mg), plasma Aβ1-x remained about 40 percent below baseline even at six days, when most of the compound was washed out, Eisai’s Robert Lai reported. All doses of E2609 had acceptable tolerability with no severe adverse events, and the safety profile was similar between young and old participants. In preclinical studies, the compound lowered Aβ levels in brain, CSF, and plasma of rats and guinea pigs, and in CSF and plasma of nonhuman primates, as shown on posters by Tatsuto Fukushima and Fiona Lucas.
Bruce Albala presented preliminary findings from Eisai’s multiple ascending-dose trial, where healthy adults ages 50-85 took the BACE inhibitor by mouth once a day for 14 days and had blood samples taken before, during, and after dosing to assess pharmacokinetics (PK) and pharmacodynamics (PD). They also had spinal taps two days before dosing and 12 hours after the final dose on day 14. Albala presented data on the 25, 50, 100, and 200 mg cohorts; the group that got the highest (400 mg) dose is still being analyzed. According to Albala, safety and tolerability looked good, with headache as the most common side effect. E2609 reaches steady state fairly quickly (five to seven days). Rising doses yield linear increases in CSF levels, indicating the compound gets into the brain and levels do not plateau, Albala said. Importantly, the inhibitor seems to be reducing amyloid production, as CSF Aβ1-x levels decreased 46 percent in the 25 mg cohort and dipped steadily further with rising doses, up to 80 percent in people taking 200 mg. CSF Aβ1-40, Aβx-40, Aβ1-42, and Aβx-42 levels all dropped as well. “These preliminary results suggest E2609 is a potent BACE1 inhibitor that results in consistent, sustained reduction of human Aβ1-x,” Albala said. Also this May, Eisai started dipping its toes into AD by recruiting MCI patients who have a confirmed biomarker for amyloid-β into a single-dose, single-center Phase 1 study.
For its part, Merck presented a slew of posters detailing preclinical and Phase 1 studies of its BACE1 inhibitors. Its lead compound (MK-8931) held its own in Phase 1, leaving some to wonder why it was not included in the talk session featuring Lilly’s and Eisai’s compounds. Merck enrolled a total of 88 healthy young adults—40 for a rising single-dose study in Belgium, 48 for a rising multiple-dose study at a U.S. site—taking blood and CSF samples at baseline, and every two hours from final dosing to 36 hours after. Indeed, “it is fairly intensive for the subjects, and we are very grateful they volunteer. We have to use a much larger needle than used for standard lumbar punctures,” Merck’s Mark Forman told Alzforum, noting that the use of larger needles increases the frequency of side effects such as headache and back pain. His poster, and another by Jack Tseng, claimed the compound has good PK and PD properties, showing dose-proportional and consistent responses among subjects at doses ranging from 2.5 to 550 mg. Maria Michener presented a cisterna magna-ported rhesus monkey model that enables more detailed PK/PD assessments because researchers can do serial CSF and plasma sampling over the course of weeks or months (see ARF ICAD story and ARF SfN story for more on this primate model).
In Phase 1, a single dose (100 or 500 mg) of the Merck inhibitor sent CSF Aβ40 and Aβ42 down more than 90 percent. In the multiple-dose study, CSF Aβ dropped 50 and 80 percent with 10 and 40 mg doses, respectively, while higher doses achieved more than 90 percent Aβ suppression. Furthermore, the compound’s 20-hour half-life makes it ideal for once-daily dosing, Forman said. A poster by K. Christopher Min showed the compound behaving similarly in a PK/PD study of healthy Japanese subjects.
According to a mathematical model developed by Huub Jan Kleijn, Julie Stone, and other company scientists, more than 90 percent of subjects can expect their CSF Aβ levels to be halved with a 12 mg dose of the Merck inhibitor. Doses around 30-40 mg should suppress Aβ by 75 percent, the model predicts. Population modeling on a poster by Lei Ma suggests that age, race, and body weight should have modest effects on MK-8931’s PK properties. Data from this analysis will help the scientists select doses for future Phase 2 trials. The company has done a small pharmacodynamics study in 30 AD patients to see if levels of Aβ reduction predicted by the model hold up in real life. Participants received 12 or 40 mg of the compound, or a higher dose (60 mg) for safety purposes. Dosing for this study was finished in June, and additional trials in AD patients are planned for later this year, Forman told Alzforum.
As further proof of principle, Merck scientist Lynn Hyde presented two posters showing that a preclinical BACE inhibitor (MBI-3) curbs amyloid buildup in the highly aggressive TgCRND8 mouse model of AD. The compound worked in preventive and therapeutic paradigms—that is, 11-week treatment of eight-week-old mice without plaques, or 18-week treatment of six-month-old mice with plaques. Treated TgCRND8 mice did not develop the phenotypes seen in BACE1 knockout mice, i.e., reduced prepulse inhibition, peripheral nerve hypomyelination, and poor navigation in the Morris water maze (ARF related news story on Savonenko et al., 2008). This suggests to the Merck scientists that these might be primarily developmental issues. Some of these data were presented at a Keystone symposium earlier this year (see ARF related conference story).
This movement on the BACE inhibition front has come with serendipitous timing. Days before the AAIC got underway, scientists reported in Nature the discovery of a protective mutation (A673T) near APP’s BACE cleavage site, which reduced Aβ production by 40 percent in human cell lines and lowered sporadic AD risk (ARF related news story on Jonsson et al., 2012). This mutation affects the same APP site as a different mutation that increases Aβ production and causes familial AD (A673V). “That is almost incontrovertible evidence that Aβ plays a critical, early role in AD,” Vassar told Alzforum. “You increase it, people get AD. You decrease it, people are protected. Genetically, it goes both ways.”
Still, BACE1 inhibitors have a long road ahead, as the field awaits data on clinical efficacy and possible side effects. On the former, some scientists fear the compounds may do little for people already showing symptoms. “If [BACE inhibitors] are tested in patients with MCI or early AD, this may be too late to show clinical efficacy,” noted Stefan Lichtenthaler of the German Center for Neurodegenerative Diseases, Munich, Germany, in an e-mail to Alzforum. “My guess is that these compounds would do better as a preventive drug, not as a therapeutic. But let’s see.” Pfizer announced last week that one of its highly anticipated Phase 3 trials of intravenous bapineuzumab showed no benefit in ApoE4 carriers with mild to moderate AD (see ARF related news story).
Weihong Song of the University of British Columbia, Canada, stressed the need to look for potential side effects. “No matter how well your compound can inhibit BACE … does it inhibit other things? That is a major question,” he said in an Alzforum phone interview after co-chairing the BACE therapeutics session with Lichtenthaler at AAIC. Recent work by Lichtenthaler and colleagues, and by Bart de Strooper’s group at the University of Leuven, Belgium, reveals additional BACE1 substrates. The work suggests that the secretase could play a role in forming connections in the brain (ARF related news story on Kuhn et al., 2012 and Zhou et al., 2012). This fits with research by Vassar and colleagues showing axon guidance defects in BACE1 knockout mice (Rajapaksha et al., 2011). “There are some mild wiring problems in the mouse olfactory system, and we are concerned these may be more general phenomena. I don’t know how serious it will be in humans,” Vassar said. “It’s not a red light for BACE inhibitors. It’s just a note of caution as we move forward with these compounds in clinical trials.”—Esther Landhuis.
You are a neurologist, and in comes a man in his fifties. He bumps into the doorframe before sitting down on the armrest of his chair, awkwardly trying to position his body in the seat. He can’t find the glass of water you place in front of him. Then he engages you in a perfectly coherent conversation about current events. Does this person have Alzheimer’s disease?
He may—sort of. But that’s not nearly the whole answer. Indeed, according to research clinicians who particularly care about patients like him, it is time to raise the profile of his peculiar and poorly understood condition. Two days before the official start of the Alzheimer’s Association International Conference held 14-19 July 2012 in Vancouver, Canada, Sebastian Crutch, Jonathan Schott, and Nick Fox of University College London, UK, together with Gil Rabinovici of the University of California, San Francisco, called the first in-person gathering of a newly formed working group on posterior cortical atrophy (PCA). The U.S. Alzheimer’s Association and Alzheimer's Research UK supported the meeting. The gathering drew a quorum of a larger group that had been forming over e-mail in the months before. More than 30 research clinicians from 20 centers in Britain, Canada, Australia, France, Germany, Holland, Italy, Spain, and the U.S. decided to move past their own idiosyncratic ways of defining PCA and see if they can accomplish more by working together. Each participating center has one to five dozen cases in its care.
The group’s to-do list is long. Besides pinning down how rare this neurodegenerative syndrome truly is, they aim to state a consensus about its core symptoms and boundaries to other diseases, and to develop diagnostic aids to be shared with the neurology community at large. They also agreed to pool samples for a genetic study of PCA’s peculiar features, and to do the groundwork for therapeutic trials specifically for these patients. “PCA is under-recognized and hugely untapped for research. It is strikingly different from typical AD,” said Schott.
PCA is a degenerative syndrome that affects the back of the brain. Some neurologists view PCA merely as atypical AD, where the disease’s amyloid deposits are distributed similarly to typical AD but its neurofibrillary tangles form more focally in occipital regions. In practice, however, PCA is essentially a clinical syndrome based on a "gestalt," as neurologists like to call it, of a collection of problems centered around visuospatial processing rather than memory.
Many patients’ initial symptoms largely fit that pattern. They cannot see objects they know are in front of them. They cannot construct in their mind’s eye a map of where things are relative to each other. For example, blind people can use a mobile phone because they retain a mental image of the keypad, but people with PCA lose that internal spatial representation. Sometimes static objects to them seem to be moving. Some symptoms are counterintuitive. One patient in Fox’s clinic, for example, recounted being unable to read the title of his daily newspaper when holding it in front of him on the train ride to work, while reading the small print just fine. Paradoxically, when looking down the train carriage, he could read the title on the same newspaper in the hands of a fellow commuter.
The symptoms can make patients disabled in daily life. Reading, writing, and calculation become difficult. People have to give up driving because they bump into things and misjudge the speed of other cars. They need help with mundane things such as finding light switches, putting a key into a lock, or picking out the can opener amid other gadgets in the drawer. “With a little help they can do almost everything; without help they can’t do anything,” Crutch recalled a carer saying.
At the same time, their memories are relatively intact, and most speak with acute insight into their condition. The British author Terry Pratchett, who eloquently raised awareness of AD through his own case, has a diagnosis of PCA. Even The Man Who Mistook His Wife for a Hat, from Oliver Sacks’ famous book, has since been speculated to have suffered from PCA. If it is ever appropriate to compare devastating diseases, PCA may be the slightly lesser evil than typical AD, and Pratchett, for his part, has said as much. “In our support group, PCA carers feel like the patient is still the person they have always known,” Crutch said.
Yet it’s important for clinical centers to develop support tailored specifically to PCA. People with this disease feel poorly served by traditional AD support groups. That is partly because they are younger and face different challenges, and partly because the activities offered in AD day centers—puzzles, large-font books—are visual and thus “actively unhelpful for people with PCA,” Crutch said. The UCL group runs regional support groups for some 200 patients with PCA in the UK and has a detailed website with information and patient videos.
The disease progresses slowly in some people and rapidly in others, but progress it does, and after some years, most patients do become memory-impaired and decline more broadly as their pathology spreads across the cortex.
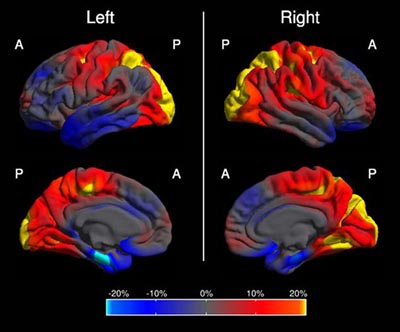
This image shows in which regions of the brain the cortex is abnormally thin in people with PCA compared to people with AD. The color scale represents the magnitude of the difference in cortical thickness; left and right refer to the brain's hemispheres. Yellow and red represent more cortical thinning in PCA (occipital and parietal cortex); blue represents more cortical thinning in AD (mediotemporal, entorhinal, frontal region). Summary image from 48 PCA and 30 AD cases, derived from Figure 1 in Lehmann et al., 2011.
How common is PCA? Are the researchers starting up a big effort for a smattering of cases? Probably not, Fox said. Determining the true prevalence and incidence of PCA requires that consistent diagnostic criteria be in place. Specialist centers have reported that 5 to 10 percent of their referrals may fit the picture, more among early onset cases. Anecdotally, when a research proposal Fox’s group had submitted to the UK Alzheimer’s Society was being discussed in a meeting with caregivers, the question arose whether this was too rare to warrant funding. “Suddenly, every layperson in the room said, ‘My husband has problems like that,’ or ‘The day center my wife goes to has someone like that,’” Fox said. From a pathology perspective, scientists know that some 25 percent of AD is atypical in terms of the distribution particularly of tangles. “We do not know what proportion of that is PCA. But even if it’s 5 percent, that is a lot of people,” Crutch said. In the U.S., that number would come to about 60,000.
That said, getting a PCA diagnosis can be a multiyear ordeal involving multiple visits to the ophthalmologist and prescription glasses until it’s clear the problem does not lie with the eyes, said Neil Graff-Radford of the Mayo Clinic, Jacksonville, Florida. Primary care providers may not see a PCA case in years, and general neurologists dealing mainly with older patients may not always recognize it. Because the symptoms are so unusual, some patients have been dismissed as malingering, Crutch said.
PCA is not new to the research community. Already in 1902, the Czech psychiatrist Arnold Pick mentioned a woman’s inability to see and grab a lit candle held in front of her face in his essay “Ueber eine eigenthuemliche sehstoerung senile dementer,” Jahrbuecher f. Psychiatrie u. Neurol., see excerpt. Several attempts at defining criteria exist. The neurologist Frank Benson at the University of California, Los Angeles, coined the term while describing the pattern of symptoms in five patients (Benson et al., 1988). Later, Mario Mendez, also of UCLA, proposed clinical diagnostic criteria arguing that PCA is its own syndrome, not just AD with visual symptoms (Mendez et al., 2002). Mendez was at the AAIC pre-meeting. Similar criteria came in 2004 from researchers at the Mayo Clinic in Rochester, including David Tang-Wai and Brad Boeve, who were both there as well (Tang-Wai et al., 2004). More recently, the International Working Group criteria (Dubois et al, 2007; Dubois et al., 2010), represented at the gathering by Bruno Dubois and Philip Scheltens, include PCA as an atypical variant of AD; they recommend diagnosis based on its core symptoms and biomarker evidence. These different sets of criteria are similar and complementary, but the group agreed that they lack detail and an agreed-upon grounding in biomarker findings.
There is a real need to collaborate, the group agreed. One problem is that, when reading another group’s papers about PCA or visuospatial variant AD, “one never knows for sure if these are the same types of patients as those at your own center,” said Fox. “We need to agree whom we are talking about.” Some research centers define PCA narrowly around its core visuospatial symptoms. Others also consider as potential PCA other cases that at first present with typically parietal symptoms such as difficulty with calculation or complex hand movements. These people may develop visual problems only years later. “They are in a diagnostic no man's land,” Crutch said. “If we want to understand AD as a whole, we need to grapple with the phenotypic continuum. PCA is not just a discrete subset. There is really no clear water between PCA and typical AD,” Crutch said.
Confused? Adding another twist to the diagnostic riddle is the fact that PCA is most often due to AD pathology, but not always. Its clinical symptoms occasionally show up as a consequence of other underlying diseases such as corticobasal degeneration, a tauopathy, or dementia with Lewy bodies. “The clinical syndromes are just constructs to describe the folks who come to see us, to help us categorize their problems,” Crutch said.
The limitations of the current clinical criteria make it difficult to replicate data that are beginning to come out on the neurobiology, biomarkers, and even immunological characteristics of PCA (e.g., Dorothée et al., 2012). They also hold back the design of new research.
Take genetics. Scientists want to know the genetic reasons for why some people get atrophy in the posterior cortex, why some get it early, and why some progress slowly and others quickly. At AAIC, Schott presented a poster on a small pilot genetic study of 58 people with PCA who are being seen at University College London, compared with 1,217 controls. Checking the loci of the AlzGene Top Results, the scientists found a different profile of effect size, even direction of effect in some cases, than these genes show in AD overall. In particular, this small study hints that in PCA, the gene ABCA7 may exert as high a risk as does ApoE. In AlzGene, ABCA7 occupies rank 4 to ApoE’s rank of 1.
The scientists need to replicate this finding with a larger group of patients, which would also allow for the possibility of identifying new genetic risks in GWAS. This requires pooling samples among the research groups working with PCA patients. Moreover, a tight definition of diagnostic criteria is important for genetic research. For example, the ABCA7 signal is stronger within a stringently defined subgroup of samples from people with core symptoms of PCA, and weaker in a larger group also including people whose symptoms place them toward the syndrome’s outer edges.
Or take clinical trials. Without clarity on exactly how to delineate PCA, how are trialists to define who should be allowed to participate in therapeutic studies? People with PCA are acutely aware of their disease and motivated to join trials, said Crutch, but there is no consistent approach at present. Some AD trials enroll people with PCA, as their criteria require no distinction among AD variants, but the protocol does not provide for their different phenotype. In those trials, PCA patients may create noise in the data. Some of the tests in those trials—visual memory, trail-making etc.—depend on skills PCA patients perform poorly to begin with because they cannot see the test properly. In contrast, they may ace auditory-based memory tests throughout the trial. Other trials exclude patients with PCA because they score too high on the MMSE, which is verbally driven.
“It seems unfair to exclude them because, based on their pathology, they are as likely to respond to the study medication as any AD patient. But where they are in trials, they fail tests for different reasons,” said Crutch. With a uniform definition of what PCA is, trialists could develop a subgroup analysis for PCA patients or develop treatment studies specifically for them, Crutch said.
Currently, there are no trials of disease-modifying compounds for PCA. A trial of donepezil in people with PCA suggested that these drugs are as modestly effective in them as in all people with AD, Crutch said. Even so, many clinicians do not prescribe these drugs to people with PCA in part because they often score well on the MMSE.
At their Vancouver meeting, the scientists decided to work toward crafting a consensus statement by the leading groups around the world on what they all agree constitutes the core and what the boundaries are of PCA. Importantly, they aim to integrate fluid biomarkers and brain imaging findings formally into the criteria for PCA. In particular, MRI and FDG-PET appear promising for the clinical syndrome of PCA. Amyloid and CSF biomarkers can be helpful in confirming that PCA is due to AD. They do not distinguish between PCA and typical AD since patients with both conditions have a similar, diffuse distribution of amyloid (Rosenbloom et al., 2011; Seguin et al., 2011). Amyloid plaques are known to start building up in the brain at least a decade before symptoms show, and scientists do not know whether the patterns of amyloid are different in the earliest, presymptomatic stages of PCA versus typical AD.
The scientists also brainstormed on collaborative projects. “We can make this group a platform to share our expertise and to study mechanistic questions of how PCA comes about, which is barely being done at all,” Rabinovici concluded. Emerging interest in PCA was already apparent throughout the main AAIC conference, with a string of presentations primarily on its brain imaging and clinical characteristics.—Gabrielle Strobel.
Most researchers agree that clinical dementia papers need reporting standards, especially when it comes to diagnostic tests. Say you are interested in this question: How well does a given biomarker predict Alzheimer’s disease (AD), or distinguish one form of dementia from another? Researchers wanting to do meta-analyses of the literature to answer questions such as these find it nearly impossible to determine the field’s knowledge because individual papers vary greatly in what and how they report. Enter STARDdem, which stands for “standards for reporting studies of diagnostic accuracy in dementia.” STARDdem is an initiative by the nonprofit Cochrane Dementia and Cognitive Improvement Group in Oxford, UK, to guide researchers in exactly how to report on diagnostic tests in the field of dementia. Compliance would help ensure that researchers control all the necessary factors and, in essence, make studies more meta-analyzable by the standards of evidence-based medicine. At the Alzheimer’s Association International Conference, held 14-19 July 2012 in Vancouver, Canada, Cochrane researchers introduced a STARDdem working document and invited the field to provide feedback during an open comment period lasting until August 31.
“There is a recognition that the literature at the moment needs a degree of standardization,” said Rupert McShane, University of Oxford, UK, who led the study. McShane heads the Cochrane dementia group, which reviews studies in the prevention, treatment, and management of cognitive impairment. “Our hope is that the STARDdem recommendations will be widely accepted and implemented.”
Modeled after the Consolidated Standards of Reporting Trials (CONSORT) project for randomized clinical trials reports (see Moher et al., 2001), the original STARD document aimed to standardize reporting in general studies of diagnostic accuracy (see Bossuyt et al., 2003). However, dementia researchers have been lax about putting its principles into practice, in part because generic standards do not apply to the unique reporting needs of the dementia field. “We are trying to make it easier for people in this field to use those criteria and understand what they mean,” said Leon Flicker, Western Australian Centre for Health & Ageing, Crawley. Flicker presented the document, now open for public comment, at the conference.
STARD is intended for studies that report the sensitivity or specificity of a given dementia test and hold that test up to the “gold standard” of AD diagnosis—usually autopsy confirmation or conversion from MCI to AD. The gold standards come some time after the diagnostic test, and this constitutes one aspect of the dementia field’s unique needs. STARDdem gives guidance on how to write up those studies, defining which aspects need to be reported. For instance, it recommends that studies report the reference standard and cite the study that validates it. It also details how to report missing data, divulge patient exclusion criteria, and include reasons for dropout, among other things. The guidelines intend to ensure that researchers report their findings thoroughly, but also that they think about and control all the important factors, said McShane. Without proper adherence to the guidelines, readers don’t always know how old a study population is or if results are from patients in a memory clinic versus the general population. “There’s rather a lot of scope in the literature for bias to creep in because the reporting suggestions of STARDdem aren't being applied,” said McShane. There are some rare exceptions; For example, a widely cited study comparing CSF diagnostic results across centers did use STARDdem criteria (Mattsson et al., 2010).
The STARDdem draft will stay open for public comment until 31 August 2012. After that, the Cochrane scientists will compile the comments and prepare a new draft, to be presented at the Clinical Trials Conference on Alzheimer’s Disease (CTAD) this October in Monte Carlo, Monaco. Soon after that, the authors hope to publish the guidelines in a scientific journal. It will then depend on journal editors and reviewers to adopt these measures and require compliance to assure widespread adoption by research groups across the field, said McShane.
Standardization is becoming especially important for biomarker studies. These are exploding in the literature and becoming increasingly important for AD diagnosis, said Henrik Zetterberg of Sahlgrenska University Hospital in Mölndal, Sweden. However, Zetterberg considered the first STARDdem draft to be insufficiently informed in terms of biomarker reporting. Zetterberg has critiqued the draft online and stressed that others should follow his example. “I think it is very important that specialists read and comment on the draft to make the first public version as good as possible,” he told Alzforum (see full comment below).
McShane’s team plans to conduct 15 literature reviews on potential diagnostic tests by September of 2013—about half on biomarker tests and half on cognitive tests for dementia. Craig Ritchie, Imperial College London, also in the Cochrane dementia group, presented a poster at the conference on the first of these reviews. He claimed that, based on the 13 studies in the literature that met his review criteria, cerebrospinal fluid (CSF) Aβ42 is neither sensitive nor specific enough to be confidently used as a diagnostic test for progression from mild cognitive impairment to AD. The poster’s conclusion met with considerable skepticism from Alzheimer’s scientists; however, AD scientists do agree that the literature for CSF tests needs to be better standardized.
“The field is at a place now where the next logical step is to bring biomarkers into the clinical realm,” said Anne Fagan, Washington University School of Medicine, St. Louis, Missouri. Before that happens, several committees will decide when and for whom such testing would be appropriate, and they will use published studies to determine those answers. While the STARDdem will not improve previous studies, it may help standardize future ones. For example, it would be helpful if authors provided simple definitions for "cognitively normal," or other qualifiers that are often neglected, to enhance comparability. “I think there has to be a common ground,” she told Alzforum.
Separately, another poster pointed to a perhaps even greater need for standardization, and that is in the still-emerging field of blood-based biomarkers. Unlike CSF, the plasma field has not begun to converge around a few markers that generate similar results among centers and studies; it is considered wide open for discovery. Andrew Watt, from the lab of Kevin Barnham at the University of Melbourne, Australia, reviewed 87 blood biomarker papers. He found no uniform way to collect and store the Aβ samples. Since the biomarker measures vary depending on the time of day samples were collected, speed of centrifugation, storage temperature, and other factors, the field really needs a standard way to collect and analyze samples and report its procedures so that studies become comparable, he told Alzforum. In toto, scientists called for a combination of quality control, assay, and sample handling standardization on the one hand, and reporting standards as developed by STARDdem on the other, to move dementia diagnosis to the next level.—Gwyneth Dickey Zakaib.
Scientists agree that Alzheimer’s disease ravages the hippocampus, making the volume of this small brain region a key marker for AD clinical trials. But as to precisely where the hippocampus starts and stops on a magnetic resonance image, researchers have little consensus. One tracing protocol can delineate a hippocampal volume more than twice as big as another method, said Giovanni Frisoni, of IRCCS Fatebenefratelli in Brescia, Italy, in a presentation at the Alzheimer’s Association International Conference, held 14-19 July 2012 in Vancouver, Canada. The Alzheimer’s Disease Neuroimaging Initiative (ADNI) has standardized how radiologists should take the images. Now, another collaborative team is stepping in where ADNI left off. The collaboration for A Harmonized Protocol for Hippocampal Volumetry, which led a lively discussion at the meeting, is midway toward its goal of developing a hippocampal trace customized for Alzheimer’s. They have consulted experts to combine disparate methods into one grand, unified protocol they hope everyone will use, making it easy to compare data among different research groups.
“Eventually, in the literature, you will be able to compare apples to apples,” said Simon Duchesne of Université Laval in Québec City, Canada, one of the presenters at the 18 July discussion. Duchesne and principal investigators Frisoni and Clifford Jack, of the Mayo Clinic in Rochester, Minnesota, envision a system whereby wannabe hippocampal tracers the world over can study the protocol, hone their skills on training images, and pass a test to obtain certification. The international project is a joint effort between ADNI and the European Alzheimer’s Disease Consortium, and they are using ADNI images as test cases.
There is no shortage of interest in the new protocol. At the Vancouver meeting, in the sixth such discussion, attendance had quadrupled since the first, Frisoni told Alzforum. The talk grew spirited as researchers debated how best to validate the tracing method. “It is critically important to get standardized protocols,” Laurie Ryan, AD clinical trials coordinator at the National Institute on Aging in Bethesda, Maryland, told Alzforum. “Being able to compare across studies is invaluable.”
The challenge is that defining the precise edge where the hippocampus meets other brain tissue is far from straightforward. It is “painstaking” work, Frisoni noted, and the researchers found 40 different ways to do it in the scientific literature. The project leaders based their plan on the 12 most commonly used methods and consulted 16 expert panelists to define the hippocampus based on four main areas.
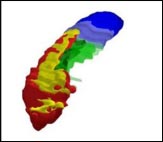
Researchers have defined the most informative hippocampal boundaries for AD, including a minimum hippocampus (red), tail (blue), subiculum (green), and alveus/fimbria (yellow). Image courtesy of Marina Boccardi, Brescia, Italy
The minimum hippocampus (red) is the undisputed hippocampal tissue; every protocol included this section. The tail segment (blue) includes everything behind the minimum hippocampus, which some tracing methods incorporated and others did not. The area called the subiculum (green), similarly, represents differences in how the top 12 protocols delineated the medial edge of the hippocampus. Finally, the researchers included white matter in the form of the alveus/fimbria (yellow), linking the hippocampus to other structures. This white matter is the not “hippocampus proper,” nor is it particularly informative for Alzheimer’s disease, noted project coordinator Marina Boccardi, who also works at the IRCCS in Brescia. However, excluding the white matter completely made it too difficult for tracers to define the hippocampal edge, so they settled on including just the part directly adjacent to the hippocampus.
The resulting hippocampal outline does not directly correspond to the brain’s anatomy. Instead, the researchers wanted to define the region that is most informative, and most reliable to trace, for studies of Alzheimer’s disease. Together, the four regions include all parts of the hippocampus affected by AD.
The current protocol runs 23 pages, with as many figures to illustrate the landmarks that define each region. Using those instructions, Boccardi’s team of five “master tracers” has outlined hippocampi with a correlation of 0.95 among them. This is an “almost perfect” match, Frisoni and Boccardi chorused in a discussion with Alzforum. Such accordance has never before been achieved with manual tracing, Frisoni said; a correlation of 0.8-0.9 is typically considered acceptable.
The next step, Boccardi said, is to validate the evolving protocol with more tracers. The protocol and images are also available to beta users who submit proposals to collaborate with the project. The team hopes to release the final version of the protocol in a year. Not only will the instructions help human tracers align their efforts, but also, researchers writing automated tracing algorithms can take advantage. Frisoni expects the protocol will be built into computerized tracing; this would be the project’s most important utility, he told Alzforum.
Scientists will be able to trace new images according to the standard, as well as go back and retrace old scans to match. With all tracers chiming in on the same tune, researchers should be better able to compare hippocampal volume across any set of drug trials, multiplying the data available to study the disease and medications’ effects.—Amber Dance.
At this year’s Alzheimer’s Association International Conference, 14-19 July 2012, in sunny Vancouver, Canada, some sessions unfolded, somewhat lonesomely, in large, sparsely populated lecture halls. This could not have been more different for a session titled "Collaboration for Alzheimer’s Prevention: Common Issues Across Presymptomatic Treatment Trials." People streamed into the room long after every seat was filled, and the crowd standing around them grew so large that fire safety rules forced closure of the room, resulting in dozens of conference attendees being turned away. For those who were shut out, here is a summary of what went on inside. First, a new group called CAP introduced itself, then three prevention initiatives gave a brief update and stood for extended discussion with the audience.
The ongoing prevention initiatives—A4, DIAN, and API—are separate, but interconnected, long-term projects. Their leaders have formed an umbrella group they call Collaboration for Alzheimer’s Prevention (CAP). "Why another acronym?" a consortia-weary reader might well ask. In short, CAP exists as a forum for A4, DIAN, and API to maintain a regular dialogue as they plan and implement their preclinical treatment trials. One goal is to avoid duplicating effort on their part and on the part of the many outside partners necessary to pull off public-private trials. Another is to ensure that the initiatives find regulatory solutions jointly and construct their trials in such a way that as many data as possible can be compared and shared with the field at large. While each initiative is unique (e.g., in which population it enrolls and in how it negotiates with pharmaceutical companies), all three have much in common, and CAP exists to exploit those synergies.
Who is in CAP? For the Anti-Amyloid Treatment in Asymptomatic AD (A4) Trial, members include Paul Aisen of the University of California, San Diego, and Reisa Sperling of Harvard Medical School; for the Dominantly Inherited Alzheimer Network (DIAN), John Morris, Randy Bateman, and Anna Santacruz of Washington University, St. Louis, Missouri; for the Alzheimer’s Prevention Initiative, Eric Reiman, Pierre Tariot, and Jessica Langbaum of the Banner Alzheimer’s Institute, Phoenix, Arizona. Working together is sometimes easier said than done; hence, facilitating the necessary conversations are Neil Buckholtz and Laurie Ryan of the National Institute on Aging; Maria Carrillo of the Alzheimer’s Association; Rusty Katz of the Food and Drug Administration; and Stacie Weninger of the Fidelity Biosciences Research Initiative.
CAP members meet several times a year to brief each other and work out points of collaboration. For example, they have been comparing notes while analyzing which psychometric tests might best measure drug effect in asymptomatic trial participants. API’s original work to look for composites in longitudinal cohorts has inspired A4 and now DIAN to do the same. In another example of collaboration under CAP, DIAN will assist in the selection and training of U.S. sites, most likely including some DIAN sites, for the first API trial of crenezumab in preclinical mutation carriers. This is done in order to distribute the burden, risk, and potential benefit of this trial between Colombia and the U.S. The Colombian regulatory authority INVIMA had indicated that it would prefer joint trial participation, and DIAN agreed to help out. Finally, to avoid measurement variability in fluid biomarkers, CSF samples from both DIAN and API will be performed in the laboratory of Anne Fagan at Washington University, St. Louis, Missouri.
The joint conversations have led to one success already. Regulators have indicated that preclinical treatment trials using a single cognitive composite and biomarkers may be able to be considered for review as registration studies in these presymptomatic populations. Typically, regulators require a co-primary, including a functional or clinical outcome.
Where does each initiative stand at this point? For A4, Aisen cited multiple longitudinal cohort studies that are showing decline among initially cognitively normal people with brain amyloid. Essentially, among healthy controls without a memory concern, the trajectories split apart when those with and without brain amyloid are analyzed separately. “This suggests that we can conduct a therapeutic trial in an asymptomatic population with amyloid, targeting changes in a primary cognitive composite that is sensitive at this period and in atrophy,” Aisen said. The A4 leaders have proposed such a trial in people 70 years of age and older with a biologically active anti-amyloid compound as part of a large application to the National Institute on Aging to renew the Alzheimer’s Disease Cooperative Study grant. They are hoping for a thumbs-up call this summer.
Aisen and Sperling propose to enroll 500 people per group. Besides its primary outcome, the trial would explore computerized tasks and patient-reported outcome measures as well as a range of biomarkers including functional connectivity—an imaging modality that requires no radioactive tracer and appears to show change early in the disease course. The trial includes two adaptive features. An early futility analysis will check whether the intervention is changing amyloid biomarkers, and a later interim analysis will confirm that the placebo group declines on the cognitive composite and re-estimate group sizes.
The choice of drug will happen in December, when ongoing Phase 3 immunotherapy trials have readout and more safety data on some Phase 2 drugs are available. “We are waiting for every last piece of information to make the best decision,” Aisen said.
Both DIAN and API have been covered in detail on Alzforum. In Vancouver, Randy Bateman reminded the audience that no group has yet offered a trial specifically for families with autosomal-dominant mutations, even though their participation in research enabled advances in AD research over the past three decades. DIAN started in 2008 as a network to support clinical trials, and has since then produced a trajectory model of presymptomatic biomarker change based to date on cross-sectional data (see ARF related news story; see also also upcoming companion AAIC story). DIAN has resubmitted a proposal to the NIA to partially fund a biomarker trial testing simultaneously three different drugs against a pooled placebo group for two years, enrolling 40 per group who are up to 15 years younger than their affected parents' age at onset. “We think there is excellent power to detect reported effect sizes on the individual biomarkers,” Bateman said.
This trial, too, has adaptive features. If one or more drug(s) work, DIAN plans to extend the study three more years to a cognitive endpoint trial using a composite. If not, then a new generation of drugs can be started on a biomarker trial. Both scenarios can happen simultaneously, provided DIAN’s expansion efforts enroll additional participants. Both DIAN and API have created registries in which prospective participants can express their interest to join a treatment study (see DIAN Expanded Registry; Alzheimer’s Prevention Registry). Bateman noted that, contrary to conventional wisdom, large numbers of autosomal-dominant AD populations are now being found, in part because DIAN is becoming widely known internationally.
For his part, Reiman noted that scientists involved in CAP use the term "prevention trial" loosely. The FDA has made clear it would not grant a claim of prevention without proof that the disease has been truly prevented for the rest of a person’s life. Therefore, the proper term is "preclinical AD treatment." API has received trial funding, made its drug choice (see ARF related news story), and is preparing to get the trial going in the first half of 2013 in people within 15 years of their affected parents' age of onset. This trial, too, foresees an interim analysis to decide whether biomarker outcomes warrant continuation of treatment. For a panel discussion with the audience, see Part 2 of this series.—Gabrielle Strobel.
This is Part 1 of a two-part series. See also Part 2.
No Available Comments
At the Alzheimer’s Association International Conference, held 14-19 July 2012, leaders of three ongoing preclinical treatment initiatives answered questions from a packed audience and from moderator Howard Feldman of the University of British Columbia in AAIC’s Canadian host city of Vancouver. Panelists were Paul Aisen of the University of California, San Diego; Reisa Sperling of Harvard Medical School; Randy Bateman of Washington University, St. Louis, Missouri; and Eric Reiman, Pierre Tariot, and Jessica Langbaum of the Banner Alzheimer’s Institute in Phoenix, Arizona.
Feldman: You have shown us a variety of different trial designs and sample sizes. Tell us why you think your design is best and what a reasonable effect size is.
Sperling: For A4 we target a 25 to 30 percent effect size, partly because that is what is known from previous trials. We would love to achieve 50 percent but have not seen that yet in AD therapeutic studies. We need better drugs for that. We chose a large group size because we do not know the exact rate of progression in our population, so we built in room for that.
Bateman: We have 40 mutation carriers in each arm, so the comparison is between 40 carriers on drug A versus 40 carriers on placebo. Be aware that we are talking about apples and oranges, because our trials use biomarker effect sizes, and A4 and API use clinical effect sizes.
That said, for example, for brain amyloid deposition, we are not expecting a 25 percent slowing of amyloid accumulation. We expect to stop accumulation or reverse it. Our points of reference are the published bapineuzumab and gantenerumab effect sizes on amyloid PET and CSF (Rinne et al., 2010; Blennow et al., 2012; Ostrowitzki et al., 2011).
Tariot: The reason so many people are in the room today is that there is a recognition that we are in uncharted space. My point is: This is unprecedented, so we are making assumptions and have to be guided by the data.
Feldman: Each of you will measure cognition in some way, but you all do it a bit differently. Each of you uses a different composite outcome.
Langbaum: The composite endpoints are actually very similar. I think the differences between the API and A4 primary endpoints will be quite minor. We found ours to be most sensitive to presymptomatic longitudinal decline in data from the Colombian Antioquia cohort and the Rush Memory and Aging Cohort and Religious Order Study. We derived two very similar composites from these two cohorts independently. A4 developed their composite using different longitudinal datasets, yet the results were similar to those found by API. In the end, certain cognitive domains are important, and each of the composites taps them in a way that is informative in the asymptomatic period. There is enough overlap between our composites to make the data very comparable.
Audience question: It’s good to bring this together through CAP. How do you address the issue of variability in the CSF measures?
Bateman: I am confident we can use these measures. Anne Fagan handles the DIAN and the API samples, so there is no lab-to-lab variability. Many of the other issues with variability and quality control of imaging and CSF are being addressed internally, with external advisors, and in separate ongoing initiatives. [Editor’s note: See ARF AAIC story.] Incidentally, that is why public funding of this trial is so important. With public funding, we can make samples available to groups that develop and validate new assays.
Audience question: How about ApoE in A4?
Sperling: The goal is to stratify but not pre-specify group sizes according to ApoE genotype.
Audience question: Have you considered combination trials?
Bateman: We gave great consideration to that. Unfortunately, there are operational and practical challenges of giving two drugs, neither of which is approved, much less from the same company. But scientifically and medically, there is a strong rationale for combination treatment.
Sperling: We would love a 2-by-2 design for an anti-amyloid antibody and a BACE1 inhibitor, or later an Aβ antibody and a tau antibody, but we cannot do that yet. That will be the A6 trial.
Audience question: Can DIAN make a pooled placebo work?
Bateman: Yes, because the primary and key secondary outcomes at this stage are the biomarker outcomes.
Tariot: On all the issues we are discussing, we need to keep in mind that there have been informal regulatory discussions but no formal approval yet from the FDA, the Colombian Ministry of Health, or ethics committees. This is new territory and new issues may come up.
Audience question: Does A4 plan to enrich for family history or subjective memory complaint?
Sperling: We will advertise for individuals who have a family history. We will admit people who report a subjective memory complaint but perform normally on the cognitive instruments.
Audience question: How about people younger than 70 who have a positive amyloid scan?
Sperling: We have discussed this. We know there are people in other studies now who are positive for amyloid. The reason we chose 70 was to be able to find a workable rate of amyloid positivity during screening, but if you are 69, we want you to be able to get in. We still need the FDA’s okay on that.
Audience question: How about treating earlier?
Aisen: We will be tracking people who were amyloid-negative at screening and seeing what happens to them. They could be in a next trial. We do not have the proper tools to do primary prevention now, but are working toward those.
Audience question: Both DIAN and API take into account that most people do not want to know their status, and the trials are powered accordingly. That is good. But enrolled people may find out their status through side effects. That needs to be said clearly.
Tariot: We will address this through informed consent. Some of the API participants in Colombia may not have much secondary education, but they are very functional and highly perceptive. They understand safety and efficacy. They are very practical.
Audience question: How are you thinking about risk-benefit? About death, severe morbidity?
Reiman: Based partly on the feedback we got from family members and external advisors, we sought an investigational agent that had evidence of target engagement (in this case, a potent amyloid-modifying effect and some diversity in the amyloid species that may be targeted) that may be associated with a reduced risk of adverse effects in healthy individuals, and safety and tolerability data (e.g., no reported microhemorrhages in the first 250 patients who have been exposed to the drug). It is important to recognize that the treatment is investigational. There is no guarantee of benefit; there is no certainty about safety or tolerability. We will have more information about safety and tolerability from ongoing trials before this preclinical trial is underway. During the selection process, we and the other committee members discussed the issue of how much efficacy (including biomarker and clinical) data would be needed before selecting a treatment. Those data are not yet available for crenezumab; however, our trial includes a 24-month interim analysis that will help in the decision to continue the trial. On managing potential risks, we believe transparent communication with the families is essential.
Tariot: We have met several hundred of these family members. Some came to our homes in Phoenix last fall. One night, 10 participants in their thirties were at our dinner table. None knows his or her genetic status. At first, some did not know each other, but by the end of dinner they looked at one another and said, “How awful that half of us will die from AD.” In other words, they know their risk. In addition to concerns for risk from the drug, also remember what the risk of AD means to them. All the standard clinical trial safety standards are being built into this trial. But they are a special population.
Bateman: The DIAN families have lived with their mutation for generations. They know what they want and what they do not want. In DIAN, there are family representatives on the steering committee and on the clinical trials committee. The family members have engaged as key partners in the design of the DIAN trial, including genetic status blinding and increasing the percent of participants receiving the active drug. The family representatives will see any toxicities and side effects as the trials unfold.
Audience question: What would be the implications of an entirely negative result of the bapineuzumab and solanezumab programs on your trials?
Aisen: Their Phase 3 programs have statistical analysis plans that give not only overall success or failure, but also information on biomarker engagement. If there is no efficacy but a strong biomarker response, would that dissuade us from using the agent? No. The clinical implications of treatment in preclinical AD are different.
Audience question: What about a negative trial with significant toxicity?
Aisen: We anticipate that the drug we choose will have risk. There needs to be an explication of risk and benefit. Some risk is acceptable. It would be a mistake to demand that people with amyloid who are cognitively normal should only be exposed to totally safe compounds.
Bateman: Paul is exactly right; the data will drive the decision. Of course, there is a balance of risk and benefit. In our case, the participants have certain risk of disease.
Tariot: There have been multiple private and public meetings here and over the last few years. Despite the aspersions some of us may sometimes cast on regulatory agencies, they have been extraordinarily helpful and willing to suggest to us what would be acceptable. Without that, our own study would not have been able to go forward.
Feldman: Clearly, the FDA wants to be partners because they see the benefit to the public. Sometimes we scientists hide behind them when, in fact, the tough decisions are in front of us.—Gabrielle Strobel.
This is Part 2 of a two-part series. See also Part 1.
No Available Comments
Researchers with eyes peeled toward prodromal Alzheimer’s disease probably see this diagram in their sleep—the one where the field’s five most validated markers trace their poignant path from normal cognition to fully developed AD. Two research groups proposed this theoretical model some years ago (see Perrin et al., 2009; Jack et al., 2010). But is it true? At least in its broad strokes, the answer seems to be yes, according to early data pouring in from biomarker studies in autosomal-dominant AD. By and large, brain amyloid, brain metabolism, atrophy, and functional connectivity data from independent analyses of the Dominantly Inherited Alzheimer Network (DIAN) and the Alzheimer’s Prevention Initiative (API) jibe with each other and confirm the sequence of preclinical biomarker changes proposed for late-onset AD. This article highlights DIAN and API neuroimaging results reported 14-19 July 2012 at the Alzheimer’s Association International Conference (AAIC) in Vancouver, Canada.
DIAN and API came onto the scene in the last few years as scientists decided to act on the desperate need and untapped research potential in people with deterministic mutations that cause early onset familial AD (see ARF DIAN series and ARF API series for background on these initiatives). These folks comprise barely one percent of AD cases, but they develop the disease at younger ages and with predictable timing. This makes them ideal for charting biomarker changes from asymptomatic stages to full-blown dementia. Early onset AD patients are eager to participate in research; to date, trials of most experimental drugs have excluded them. On that, the tide is starting to turn. The U.S. National Institutes of Health announced in May that it would help fund the first-ever AD prevention trial in at-risk adults with no outward signs of AD—namely, Colombian people with the “Paisa” (E280A) presenilin 1 mutation who are enrolled in the API registry. Slated to start in 2013, the $100 million trial will test the monoclonal antibody crenezumab (see ARF related news story; see also ARF news story). DIAN—the study of the globally scattered families whose Alzheimer’s stems from a mutation in the amyloid precursor protein (APP) or presenilin (PS) genes—is also gearing up for therapeutic trials (see ARF related news story). So is another effort called Anti-Amyloid Treatment for Asymptomatic AD or A4, which intends to enroll healthy seniors with amyloid-positive brain scans (see AAIC story; ARF Q&A).
In Vancouver, Randall Bateman of Washington University School of Medicine, St. Louis, Missouri, recapped DIAN findings reported recently in the New England Journal of Medicine (Bateman et al., 2012) and in talks and posters throughout AAIC. At the AAIC’s imaging consortium pre-meeting, WashU’s Tammie Benzinger and colleagues won the Best Poster award for their analysis of brain amyloid, glucose metabolism, and cortical atrophy in 191 DIAN participants, including 119 mutation carriers. This is a more recent, larger version of the cohort of 128 analyzed for the NEJM report. The carriers had detectable brain amyloid some 15 years in advance of symptoms, metabolic decline and hippocampal shrinkage about 10 years prior, followed by cortical thinning and disrupted functional connectivity about five years before signs of disease. “These findings were reported for DIAN across three labs and two continents, using different analytic and statistical approaches to the data,” wrote Benzinger, who leads the DIAN imaging core, in an e-mail to Alzforum. At present, these data constitute cross-sectional comparisons; longitudinal data are still being gathered.
While the pattern of the DIAN imaging data for the most part matches what scientists see in sporadic AD, it is not identical. Unexpectedly, the DIAN cohort showed presence of white matter disease in mutation carriers. This is common in people with sporadic AD and is usually attributed to comorbid conditions such as high blood pressure or diabetes. However, DIAN participants are younger and generally do not have these comorbidities. “More investigation is needed to see whether the white matter findings are a consequence of the grey matter amyloid plaques or are perhaps related to amyloid deposition in blood vessels in the DIAN cohort,” Benzinger said. An expected difference on amyloid PET imaging is early deposition in basal ganglia, which has been reported previously in some cases of autosomal-dominant but not in sporadic AD.
In a featured research session on neuroimaging for presymptomatic trials, Alex Becker of Massachusetts General Hospital, Boston, presented initial data on baseline glucose metabolism in the brains of DIAN participants. Becker’s team analyzed how metabolism differed with mutation status and age (i.e. years from expected symptom onset) in 124 people who were either cognitively normal with a Clinical Dementia Rating (CDR) score of 0, or mildly symptomatic with CDR 0.5 or 1. As expected, symptomatic mutation carriers had lower brain activity—measured with fluorodeoxyglucose (FDG) positron emission tomography (PET)—than the control group of cognitively normal non-carriers. Moreover, Becker reported, the FDG signal showed a similar regional pattern as in sporadic AD, with metabolism waning in the precuneus and other AD-vulnerable brain areas, and falling off more steeply as carriers became more impaired. When comparing only asymptomatic subjects, mutation carriers had greater metabolic decline than non-carriers in two areas—an AD-vulnerable aggregate region and the angular gyrus. The hypometabolism became more pronounced as carriers approached their family’s age of symptom onset.
Furthermore, the researchers examined functional connectivity in the brains of DIAN participants. With this imaging modality, too, they saw differences between asymptomatic mutation carriers and non-carriers. Jasmeer Chhatwal of MGH and colleagues looked at resting-state connectivity in the default-mode network in functional MRI scans of 120 DIAN participants (83 mutation carriers and 37 non-carriers from the same families). People with sporadic AD are known to have decreased connectivity in their default mode network—a group of brain areas that fire up when the mind is resting (ARF related news story on Greicius et al., 2004). Similarly, mutation carriers in the DIAN cohort had decreased connectivity throughout the default-mode network, compared to non-carriers, even if they were still asymptomatic with a CDR of 0. Also like sporadic AD, functional connectivity in DIAN subjects decreased more quickly as they approached their family’s age of symptom onset, and continued to worsen with disease progression (see image below). “We are very excited about this data,” Chhatwal told the AAIC audience. Intriguingly, it looked as though the youngest subjects actually had increased connectivity, but this needs further study with larger sample sizes, he said. At last year’s AAIC in Paris, scientists at the Banner Alzheimer’s Institute, Phoenix, Arizona, reported that among 18- to 26-year-olds enrolled in API, mutation carriers already show differences in fMRI patterns (see ARF related news story), prior to amyloid deposition measurable by PIB. In the future, Chhatwal and colleagues plan to look at functional connectivity in other brain networks besides default mode, and to analyze whether the fMRI data correlate with amyloid imaging, CSF Aβ and non-amyloid biomarkers, as well as with specific mutation types.
Adults with autosomal dominant Alzheimer’s disease mutations show decreased connectivity throughout the default-mode network (DMN) relative to non-mutation carriers, as revealed by functional magnetic resonance imaging (fMRI) brain scans. Image courtesy of Jasmeer Chhatwal, Massachusetts General Hospital
Reisa Sperling of Brigham and Women’s Hospital, Boston, is a DIAN site leader who has seen the functional MRI data from both DIAN and API cohorts. “There is remarkable convergence in the evidence that the default-mode network is dysfunctional prior to any cognitive impairment, and perhaps even prior to amyloid deposition in some subjects.” Sperling wrote in an email to Alzforum.
Now on to API data. At the Vancouver meeting, Adam Fleisher of the Banner Alzheimer’s Institute in Phoenix, Arizona, spoke about florbetapir amyloid PET, cerebrospinal fluid (CSF), and MRI biomarker changes in API participants at preclinical and clinical stages of early onset AD. Fleisher had presented some of the florbetapir data in January at the Human Amyloid Imaging Conference in Miami, Florida (see ARF related news story). On the whole, biomarker profiles in API mutation carriers looked similar to the hypothesized trajectories for sporadic AD, Fleisher wrote to Alzforum. Fibrillar Aβ deposits cropped up about 16 years before clinical symptoms, followed a few years later by CSF Aβ42 and subsequently tau changes, and then hippocampal shrinkage about seven years in advance of symptoms.
Given that adult carriers as young as 18 to 26 already appeared to have fMRI and even some subtle brain structure changes compared with non-carriers, the API scientists reached back even further in age and decided to look for changes in 9- to 17-year-old children with the Paisa PS1 mutation. Yakeel Quiroz of Boston University, Massachusetts, and Universidad de Antioquia, Colombia, and colleagues scoured MRI scans of 21 mutation carriers in this youth bracket. They measured cortical thinning in nine signature regions previously defined in patients with mild sporadic AD (Dickerson et al., 2009). This MRI signature can predict which MCI patients will convert to AD (Bakkour et al., 2009) and, in a longitudinal study, predicted which healthy seniors eventually develop AD (Dickerson et al., 2011). Even in this youth bracket, mutation carriers were distinguishable, but in a surprising way—by having a thicker cortex than age-matched non-carriers. However, the young carriers lost brain tissue more quickly and had a thinner cortex, on average, than age-matched non-carriers by the time they reached their 30s. Older, symptomatic carriers continued to show faster cortical thinning in all nine signature regions, as well as in the signature’s summary measure.
Among the AD prevention initiatives, API has the most genetically homogeneous participants, as all have the same PS1 mutation. DIAN participants have one of dozens of familial AD mutations, and the A4 cohort is heterogeneous, enrolling any asymptomatic senior with biomarker evidence of amyloid. MRI profiles may, however, differ according to FAD mutation. David Cash and Kirsi Kinnunen at University College London, U.K., analyzed structural MRI data from a local cohort of presymptomatic FAD mutation carriers, some of whom also enrolled in DIAN. Thirty-one people with PS1 mutations and 12 with APP mutations received several MRI scans about a year apart and were followed longitudinally for up to two decades. The analysis included 44 age- and sex-matched non-carriers, each scanned twice about 18 months apart. By the time symptoms appeared, mutation carriers already had higher atrophy rates than non-carriers in all structures analyzed (whole brain, ventricles, hippocampus); however, the MRI profiles were not the same in all carriers. APP mutation carriers seemed to lose tissue faster in the hippocampus, while PS1 carriers showed accelerated atrophy in the global measures of whole brain shrinkage and ventricular expansion, Cash reported on a poster.
Likewise, structural MRI profiles differed between PS1 and APP carriers in a cross-sectional analysis of 190 DIAN participants, as presented on a poster by Kinnunen. This cohort comprised 119 mutation carriers (95 PS1, 10 PS2, 14 APP) and 71 non-carriers. When the PS1 mutation carriers were studied separately, presymptomatic carriers had smaller whole-brain volumes than non-carriers. However, this finding disappeared when brain volumes from all mutation carriers were lumped together and compared with non-carriers, suggesting that individual FAD mutations may affect brain volume differently. What do these tidbits of data on different imaging modalities, age brackets, and mutations amount to at this point? “I think the data coming out on DIAN and API biomarkers looks overall great so far. The bottom line is that things seem consistent with the amyloid cascade,” noted WashU’s David Holtzman, who sits on the DIAN steering committee. “It looks very encouraging in the sense that there is quite a long window where one could initiate prevention trials.” Nick Fox of the London group agreed, but added that in terms of designing those trials, the early biomarker changes in asymptomatic FAD mutation carriers present both a challenge as well as an opportunity. “Do we aim for trials in individuals many years before symptom onset, perhaps at a stage when there is very little evidence of any irreversible changes? Or do we aim for subjects who are closer to onset age, when tracking progression and assessing effects of therapy would be easier and quicker?” he wrote to Alzforum.—Esther Landhuis.
No Available Comments
Researchers think amyloid deposition, synaptic disruption, and neuronal glucose metabolism are all somehow connected as Alzheimer's disease (AD) unfolds, but they have not yet been able to explain just how, when, and where. Members of Alexander Drzezga’s lab from the Technical University in Munich, Germany, use longitudinal multimodal imaging to tackle aspects of that question. They presented their latest findings at the Alzheimer’s Association International Conference (AAIC), held 14-19 July 2012 in Vancouver, Canada.
Stefan Förster has set out to determine how plaque deposition and metabolism (a measure of synapse dysfunction) relate to each other over time. He published earlier this year that areas of plaque deposition at a baseline scan in people with mild AD became hypometabolic by about 27 months later (see Förster et al., 2012), suggesting that plaques cause a drop in metabolism. On an AAIC poster with his latest longitudinal data, he reported that metabolism predicts plaque deposition as well.
Förster followed 15 mild AD patients for about two years using [18F]-2-deoxy-2-fluoro-D-glucose (FDG) positron emission tomography (PET) scans to show metabolic function, and Pittsburgh compound B (PIB) to detect plaques. He found that areas of relatively preserved metabolism at baseline were overrun with amyloid two years later. In addition, as in his previous work, regions with baseline amyloid were less metabolically active at follow-up. The results point to a causal triangle, where initial neuron activity is followed by amyloid deposition, which in turn leads to functional neuronal decline. “This bidirectional finding suggests that there is a causal relationship of these two biomarkers in neurodegenerative AD,” Förster told Alzforum. “I think this is the first report of such a bidirectional relationship in early AD patients.”
But what could explain hypometabolism in regions that do not have much amyloid in the first place? Researchers have previously found that network activity in AD patients goes awry, especially in the default-mode network, which is active during sleep or when the mind wanders (see ARF related news story on Sperling et al., 2009; Hedden et al., 2009; Sheline et al., 2010). Perhaps a network breakdown is at play here, too, suggested Elisabeth Klupp, who spoke about her findings at an oral presentation. “We hypothesized that hypometabolism in non-amyloid affected brain regions could result from functional disconnection from remote brain areas affected by these pathologies,” she told her audience.
To find areas of hypometabolism with little plaque, she and her colleagues used multimodal imaging in 19 AD patients. An averaged FDG-PET scan from them compared with one from 15 age-matched healthy controls revealed areas that were abnormally hypometabolic in the AD patients. By superimposing that map on an averaged PIB-PET scan from the AD patients, the researchers saw areas with reduced metabolism and no significant plaque, most prominently the left superior frontal gyrus. Hypometabolism there exceeded what the team would expect, given the amount of amyloid. This region then functioned as a “seed” to figure out which areas were normally functionally connected to it in healthy controls.
In 17 elderly healthy controls at rest, functional magnetic resonance imaging (fMRI) scans revealed that parietal areas, especially on the left side of the brain, communicate with the left superior frontal gyrus. In the AD patients, according to PIB-PET, these same areas were overrun with amyloid. The overlap suggests that a breakdown in communication between areas with too much and very little amyloid may cause the unexplained hypometabolism, suggested Klupp.
“You might have amyloid in one place inducing neuronal damage that is mirrored at the other end,” Alexander Drzezga, principal investigator on the project, told Alzforum. Drzezga compared the situation to a faulty phone connection: If the talker suddenly goes mute, the listener hangs up. Likewise, if one brain region stops sending out signals, the recipient will not receive as many, and its metabolism will drop. The data appeared in a 2012 supplement of the Journal of Nuclear Medicine.
“This study for the first time explains distant effects on glucose metabolism through connectivity to areas of high amyloid deposition,” wrote Victor Villemagne, University of Melbourne, Australia, to Alzforum in an e-mail. Previous studies have started with areas of high amyloid deposition and examined how they were functionally connected to other areas, he said. “This study reverses that paradigm, placing the seed in a region of hypometabolism and tracking which regions are connected,” he told Alzforum. “This reversal is very original, adding to our knowledge of how different regions interact which other, and how amyloid deposition in one area might affect the functioning of a distant region.”
In the future, Drzezga’s research group plans to further explore a cross-sectional and longitudinal interrelation between amyloid burden, hypometabolism, and functional disconnection. In particular, they will check longitudinally for changes in AD patients’ connectivity and see how those changes relate to areas of amyloid deposition down the road. “The ultimate goal of this research is to understand the pathophysiology of neurodegeneration in Alzheimer’s disease,” Förster told Alzforum. “With these imaging modalities, we have the unique opportunity to follow the neuropathological changes in vivo and get a signature of this disease.”—Gwyneth Dickey Zakaib.
No Available Comments
As the Alzheimer’s disease (AD) field moves closer to using genetic and biomarker data to identify people at risk, researchers are urgently trying to tackle whether and how to disclose that information to people in both routine clinical care and research settings. This past February, Alzforum published a detailed account of the issues involved and ongoing studies aimed at these goals (see ARF related news story). In Vancouver at the Alzheimer’s Association International Conference 2012, three of the featured researchers updated attendees on their projects at a plenary session dedicated to the topic.
“This move from research to clinical application is starting to occur with different biomarkers,” said Scott Roberts, University of Michigan, Ann Arbor. Chairing the session, Roberts spoke of the need to use biomarkers responsibly in research. The coming shift from research to clinic “raises the stakes in terms of trying to think about how we communicate this information to providers and patients,” he told the audience.
First up was Robert Green, Brigham and Women’s Hospital, Boston, who for a decade has been conducting research examining whether it is psychologically harmful to divulge ApoE4 carrier status, which is associated with a higher risk of AD, to cognitively normal people (see ARF related news story and ARF Live Discussion). For the most part, the answer has been, no. “We have been able to chip away at this notion that genetic information is inherently harmful,” he told the audience.
But what if a person already has mild cognitive impairment (MCI)? Having both an ApoE4 allele and MCI means a person has about a 50 percent chance of developing AD within three years (see ARF related news story), a rather more imminent prospect than the comparatively abstract concept of lifetime risk. That is why Green and colleagues are now conducting REVEAL IV. In this study they assess the risk of telling patients with MCI, ages 55-90, and their study partners whether the patient carries ApoE4. So far, the research team has preliminarily assessed only a small group of 30 patients—half of whom got their results and the other half who didn’t—to be sure the study is safe. The researchers assessed anxiety, change in health behavior, and insurance/lifestyle changes by phone one to three days after disclosure, then again at six weeks and six months. “So far we have not demonstrated robust anxiety, depression, or distress among either the subjects or their partners when they receive E4 information,” Green said. “We can start to assuage the concerns of people who felt that this was a bad idea.”
But ApoE4 status, even if you have MCI, still only imparts genetic odds. What if people already have plaque buildup in their brains? Patients are likely to consider that a more definite sign that they are on the road to Alzheimer’s. With the recent FDA approval of Amyvid (see ARF related news story) as the first of an expected handful of F18-labeled amyloid detectors for positron emission tomography (PET), many clinicians are gearing up for potential public demand of the test (see ARF related news story). “The increasing popularity and pending commercial availability of amyloid imaging tools are raising a number of questions about disclosing amyloid imaging results under various clinical scenarios,” Jennifer Lingler, University of Pittsburgh, Pennsylvania, told the audience. One question is whether knowing that one is likely developing Alzheimer’s heightens anxiety in people who already know something is wrong with their minds.
Since the PET tracer aims to help rule out AD as a cause in people with cognitive deficits, some of the first to get the test will likely be people with MCI. For that reason, Lingler’s trial involves disclosing a mock plaque status to MCI patients and their caregivers. (Patients know it is a mock trial.) For now, Lingler wants to know if information is presented in a comprehensible way. A panel of experts developed standard scripts to deliver in a disclosure session, and Lingler tried them out in 10 pairs of patients and family members. Four pairs each heard "positive" and "negative" scripts, while two received the inconclusive script.
Participants later rated how strongly they agreed or disagreed with statements such as: The session was “easy to follow,” “included the right level of detail,” etc. The feedback was generally positive, Lingler said. “Folks found the session easy to follow and the information clearly presented,” she said. The team is taking into account all feedback and additional questions participants asked, and will use them to make modifications to future scripts. They will also build in some flexibility with regard to length of the session and level of detail, depending on the participant’s preferences. Lingler’s next trial will assess outcomes of MCI patients who receive real imaging results.
Perhaps an even more contentious issue is whether doctors or researchers should reveal plaque status to cognitively normal people. While amyloid deposition in people with MCI appears highly predictive of progression to AD dementia, the research on whether cognitively normal people with plaque progress to AD is at an earlier stage. Jason Karlawish, University of Pennsylvania, Philadelphia, is part of a team tackling that question within the A4 trial (see ARF related news story), in which participants will learn their plaque status by virtue of taking part in the study, he explained. “We have great concern that it may cause despair,” said Karlawish. Within the A4 trial, he and colleagues will deliver scripts developed by amyloid imaging and genetic counseling experts to prospective A4 participants as part of counseling both before and after the amyloid test. The scripts explain what amyloid imaging is and what it means to have a positive or negative result. The researchers will check whether participants understand their amyloid status and risk. They will monitor participants’ mood throughout the study and note any changes in lifestyle, behavior, or perceived quality of life after disclosure. These results will inform future protocols that the research group will help develop, Karlawish said.
A parallel effort to issue consensus guidelines for diagnostic disclosure of biomarker status in people with MCI is afoot in Europe as part of the European Alzheimer’s Disease Consortium (EADC) (see ARF related news story). The guidelines themselves are not quite baked yet, according to Pieter Jelle Visser, University of Maastricht, the Netherlands. In the meantime, however, leaders of the initiative are outlining their thinking in a freely available editorial in a special prevention issue of the journal Biomarkers in Medicine. The authors, including Visser, advocate for shared decision-making, which entails a joint decision between both patient and provider to get biomarker results. The process will likely include thorough pre-test counseling in which the provider explains the current uncertainties in biomarker statistics, possible test outcomes and their meaning, and risks involved with getting tested.—Gwyneth Zakaib.
No Available Comments
Induced pluripotent stem cell (iPSC) models are flourishing in disease research. Though researchers cannot use them to study learning and memory the way they would in animal models, these patient-derived cells may illuminate the underlying biology of human disease at the cellular level in a way animals cannot. “By going straight into human cells, the biology more closely reflects what is happening in people,” Rick Livesey, University of Cambridge, UK, told Alzforum. Livesey was one of several researchers who highlighted the latest efforts at developing iPSC lines to advance Alzheimer’s disease (AD)-related research at this year’s Alzheimer’s Association International Conference held 14-19 July in Vancouver, Canada.
Livesey published earlier this year on an iPSC model of Down’s syndrome (DS) developed from patients’ skin cells (see Shi et al., 2012). His team took skin cells from adults with DS and generated iPSCs, which he then induced to become cortical neurons. Those neurons, which were the focus of his presentation, are a relevant model for AD research because almost two-thirds of individuals with DS get dementia by the time they are 50. People with Down’s have an extra chromosome 21, which harbors the amyloid precursor protein (APP) gene, and they develop amyloid plaques and tangles as early as their thirties.
Livesey emphasized that DS neurons take months to develop in a culture dish, about as long as they normally do in situ, meaning they probably take a close-to-natural route. The cells—mostly glutamatergic neurons and some glial cells—also acquire mature firing properties, firing synchronously, and they form synapses with each other. This is important when studying diseases of synaptic dysfunction, said Livesey. The cells display an AD-like phenotype in that they secrete more Aβ40 and Aβ42 than do undifferentiated precursor cells and skin cells. Later, at three months of age, Aβ42 forms aggregates in these neurons, and tau becomes hyperphosphorylated and gets mislocalized into the soma and dendrites.
Recently, Livesey showed the cells’ usefulness as a screening platform. Not only do they thrive in 96-well plates, but they also recapitulate known effects that drugs such as γ- and β-secretase inhibitors show in other cell and animal models. Livesey’s lab is now using these iPSC-derived neurons to screen FDA-approved drugs used in other diseases, since they are already known to be safe for human use, to see if any reduce the AD phenotypes seen in DS. “We all agree that these models need to go from being a proof of concept to actually teaching us new things,” Livesey said. In the future, Livesey plans to delete a gene at a time from chromosome 21 to see which ones rescue which aspect of the cells’ phenotype. He is collaborating with Larry Goldstein, University of California, San Diego, and John Hardy, University College London, to make iPSC models of familial AD and see how similar they are to the DS model.
Also in Vancouver, Alysson Muotri, University of California, San Diego, gave an example of how he has taken the concept of iPSCs for drug discovery a step further—in this case, for autism. Gene sequencing of a patient with sporadic autism revealed that one particular gene—TRPC6—was mutated so that expression of the calcium channel it encodes fell by half. One drug, hyperforin, isolated from the plant St. John’s wort, was already known to bind to TRPC6 and induce calcium influx. In human iPSC-derived TRPC6 mutant neurons, hyperforin stimulated calcium influx. Encouraged by that result, the researchers tried hyperforin treatment in the child whose cells they had collected for the study. While they only have a single time point so far, the child’s father reported improved behavior and the school reported better focus, Muotri claimed. The group will continue to follow that patient. “The coolest part of iPSCs is being able to move from the bench to a translational approach,” said Muotri. “The combination of genomics and iPSC studies is powerful.”
AD cell models are further behind, but researchers are working to catch up. Andrew Sproul of the New York Stem Cell Foundation (NYSCF), New York City, presented on his development of 72 iPSC lines from 12 affected and unaffected members of two families carrying presenilin mutations whose skin cells were collected in the 1980s and were recently reprogrammed into stem cells. The cholinergic cells derived subsequently from these skin cells were electrically active and, just like Livesey’s DS cells, produce more Aβ than do controls. Moving forward, NYSCF is automating the production process and creating a larger number of familial and sporadic iPSC-derived neurons from current patients to aid in the observation of biological disease processes as well as in drug safety and efficacy studies.
DS iPSC-derived cortical neurons have Aβ42-containing aggregates, shown in green. These deposits are absent from both control iPSCs and DS-derived fibroblasts. Image courtesy of Science Translational Medicine/AAAS
iPSC-derived neurons from members of a family carrying a presenilin mutation. Images courtesy of Andrew Sproul, NYSCF
Though iPSC cells hold much promise, they still have a way to go to prove their worth, said Edward Koo, University of California, San Diego, who chaired the stem cell session at which Livesey and Muotri presented. "iPSC technology has not reached its full potential yet,” he told Alzforum; hence, no one can be sure just how valuable these cells will be in furthering AD research. “In what ways they will be better than existing models remains to be seen, but from what we heard today, it's very exciting,” he said.
A recent paper highlights the potential use of iPSCs for drug discovery in other diseases. In the August 1 Science Translational Medicine, Haruhisa Inoue, Kyoto University, Japan, and colleagues report the creation of nine iPSC lines from three familial amyotrophic lateral sclerosis (ALS) patients. Each patient was heterozygous for a deleterious Tar DNA binding protein-43 (TDP-43) allele. First author Naohiro Egawa saw that, as in animal models and postmortem human tissue, motor neurons generated from these cells overexpressed TDP-43 mRNA, had cytosolic aggregates of TDP-43 protein, shortened neurites, and were more vulnerable to agents stressful to cells, such as arsenite, compared to controls.
In the ALS neurons, gene transcription relevant to RNA metabolism was perturbed. For this reason, researchers used the motor neurons to test four potential treatments known to modulate transcription in an arsenite-induced motor neuron death assay. Of those drugs, the histone acetyltransferase inhibitor anacardic acid fixed some of these neurons’ atypical characteristics. The compound quashed TDP-43 mRNA expression, reduced insoluble TDP-43, boosted cell survival in the face of arsenite treatment, and lengthened neurites. Inoue and colleagues will continue to test anacardic acid’s safety and efficacy in other ALS models. Meanwhile, this cell model could be useful for determining the disease’s underlying biology, and might aid in future drug discovery, wrote the authors.—Gwyneth Dickey Zakaib
No Available Comments
In the past three years, genomewide association studies have grown the list of Alzheimer’s risk genes from a lone big gun (ApoE4) to a collection of 10 or so that account for about a third of the genetic risk for late-onset AD. That still leaves two-thirds of LOAD heritability a mystery, but on that front, a huge meta-analysis of GWAS data is turning up additional genes, scientists said at the Alzheimer’s Association International Conference held 14-19 July 2012 in Vancouver, Canada. In other genetics news at the conference, the first-ever African-American AD GWAS suggests ApoE may not be top dog in this ethnic group, and new analyses are homing in on biological pathways underlying existing risk variants to figure out how they actually contribute to AD. For example, some data suggest that BIN1 might bind tau and boost tauopathy.
In early 2011, scientists launched the International Genomics of Alzheimer’s Project (IGAP), pooling genetic data from four consortia in the U.S., U.K., and Europe into a mega-dataset containing 8.5 million single-nucleotide polymorphisms (SNPs; see ARF related news story). Analysis of this set has confirmed that all risk genes found in prior AD GWAS are indeed robust, Gerard Schellenberg of the University of Pennsylvania School of Medicine, Philadelphia, reported at AAIC. Schellenberg heads the Alzheimer’s Disease Genetics Consortium (ADGC), the largest of the four consortia in IGAP. At the same time, the real goal of IGAP is to identify new AD risk genes—especially the rarer variants present in as few as 1 percent of the population, which would escape detection by commercial genotyping platforms. Toward this end, the scientists culled from the mega-dataset around 20,000 SNPs with p-values of less than 1.0 x 10-3 and put them onto a custom chip for a replication phase using 29,000 subjects that were not part of the mega-meta-analysis. That is a liberal cutoff, Schellenberg said, since SNPs are not typically deemed genomewide significant unless their p-values fall around or below 1.0 x 10-8. The genotyping process for this project is more than 75 percent complete, Schellenberg told Alzforum. He did not disclose the names of the new risk genes, or even how many have been identified, at AAIC, but told Alzforum the team plans to draft a paper this month.
Pathway analysis offers another way to find new risk genes. This method involves looking at a list of potentially significant genes and testing whether more of them lie in a biochemical pathway than would be expected by chance. The list includes top hits as well as SNPs in the uncertain zone that came close to genomewide significance but failed to reach the statistical cutoff. “Some of those SNPs could be real, but you don’t know which ones. However, if 5 percent of them end up in one pathway, you can start to have confidence that they may be significant,” Schellenberg said.
Peter Holmans of Cardiff University School of Medicine, U.K., has done pathway analysis of IGAP data. This analysis has not turned up new risk genes as of yet; however, it has shed some light on the biological processes that may underlie genetic susceptibility to late-onset AD. Of 9,816 pathways tested, those relating to endocytosis and cholinergic receptors contributed most strongly to late-onset AD, Holmans reported in an AAIC talk. While these pathways are not new, “it is good to have their involvement confirmed by a systematic analysis of genomewide data,” Holmans said. Even after known variants (ApoE, BIN1, PICALM, etc.) were removed, these pathways still came up strong, Holmans noted, suggesting that their involvement in AD may be more general, not restricted to existing risk genes. The analysis also uncovered several new pathways—for example, protein catabolism, and degradation through ubiquitination pathways and the proteasome.
Christiane Reitz at Columbia University College of Physicians and Surgeons, New York, reported preliminary findings from the first large GWAS on AD in African-Americans. Her team used about 5,200 samples from the ADGC cohort that had not been tested in previous GWAS. The top hit was ABCA7, which came up in recent large GWAS in Caucasians (Naj et al., 2011; Hollingworth et al., 2011). However, the new and important finding here was that ABCA7 and ApoE seem to influence AD risk equally in African-Americans. “The effect size of ABCA7 was nearly as strong as the effect size of ApoE. The reasons for this remain to be clarified,” Reitz told Alzforum in an e-mail. The preliminary analysis also uncovered other SNPs in pathways previously reported to play a role in AD: CTNNA3 in cell-cell adhesion, CAPRIN2 in synaptic plasticity, and SPG20 in endosomal trafficking.
What about existing AD risk variants that have been confirmed time and time again? Consider BIN1, for example. Beyond ApoE, it is the top genetic susceptibility locus in AD, yet scientists know little about how it contributes to the pathophysiological process. Recent work from Jean-Charles Lambert’s research group at INSERM in Lille, France, has begun to change that. In several AAIC talks and posters, Lambert and colleagues put forth the possibility that Bin1 could interact with tau and exacerbate its toxicity. In a prior analysis, the researchers saw that BIN1 was overexpressed in AD brains. When they knocked down expression of the BIN1 Drosophila orthologue (Amph) in fruit flies, they found that this suppressed tau toxicity while leaving Aβ-related phenotypes unaffected. Those findings were reported at last year’s AAIC in Paris, France (see ARF related conference story). At the Vancouver meeting, Julien Chapuis from the group extended these findings with a poster showing evidence that Bin1 interacts with tau. The proteins associated in GST pulldown assays when overexpressed in transformed human cell lines, and also in synaptosomal fractions purified from wild-type mouse brain. The researchers are now investigating the consequence of this possible interaction, asking, for instance, whether Bin1 affects tau phosphorylation or aggregation, or influences formation of paired helical filaments.
Rudy Tanzi of Massachusetts General Hospital, Charlestown, finds the data interesting because of a possible tie-in with recent work published by Susan Lindquist of the Whitehead Institute for Biomedical Research in Cambridge, Massachusetts. In that study, which included Tanzi as a coauthor, scientists searched for modifiers of Aβ toxicity using a yeast screen. Three genes that came out of that screen are related to AD GWAS hits. One encodes synaptojanin 1, which interacts with Bin1. In this study, Bin1 is in a complex that affects Aβ toxicity, while the INSERM study suggests that Bin1 affects tauopathy. “Maybe Bin1 helps bridge Aβ pathology to tau toxicity,” Tanzi said. “How tauopathy is triggered from excess accumulation of Aβ has been a black box in the AD field for some time.” Then again, if a LOAD genetic factor like BIN1 can directly influence tau toxicity without affecting Aβ pathology, “it could be a strong argument against the amyloid cascade hypothesis,” Chapuis suggested in an e-mail to Alzforum.—Esther Landhuis.
No Available Comments
At the Alzheimer’s Association International Conference held 14-19 July 2012 in Vancouver, Canada, both epidemiological and lab findings converged to point a finger at sleep in connection to cognitive decline. In particular, objective sleep measures suggesting that sleep disorders predict decline added a new layer of experimental data to older self-reported hints that how long and how well one sleeps might affect brain health later in life.
“It is unusual to have [multiple] studies coming together at the same time to say the same thing,” said Constantine Lyketsos, Johns Hopkins Bayview Medical Center, Baltimore, Maryland. “Sleep disruptions throughout the lifespan are probably accelerators of cognitive aging and risk factors for dementia,” he said.
Kristine Yaffe, University of California, San Francisco, presented her recent epidemiological findings on sleep—some published and some not. Yaffe and colleagues followed a prospective, multicenter cohort of community-dwelling women, all of whom were 65 or older and free of dementia at enrollment. Fifteen years into the study, about 3,000 of those women underwent actigraphy, whereby a watch-like device worn on the wrist measures sleep by recording movement over several days. They also underwent polysomnography, a lab-based sleep study that takes more in-depth measurements of overnight sleep patterns. Five years after those tests, the scientists checked to see who had since developed mild cognitive impairment (MCI) or dementia.
Yaffe found that several sleep-related problems raised the risk of subsequent cognitive decline. For one thing, a late shift in the women’s circadian rhythm, where their mean peak daily activity moved by two hours from early to late afternoon, was linked to an almost two-fold increased risk for developing MCI or dementia (see Tranah et al., 2011). Separately, sleep-disordered breathing (or sleep apnea) at baseline also heightened the risk of developing MCI or dementia almost twofold. This latter finding may be explained by hypoxia rather than sleep fragmentation, said Yaffe (see ARF related news story on Yaffe et al., 2011). In addition, a delay in falling asleep and less efficient sleep both increased the risk of being placed in a nursing home five years later.
“The effect is probably bidirectional, meaning disordered sleep may cause dementia at the same time that dementia causes sleep trouble. But at least we are showing that sleep problems are predicting five-year outcomes related to nursing home placement and MCI and dementia,” Yaffe said at a press briefing. The mechanisms are not yet clear, but besides hypoxia they could have to do with Aβ accumulation, she added (see Part 2). The findings matter, she said, “because sleep problems could be treatable, and intervention might delay or prevent some of the cognitive sequelae.” A next step would be to do a randomized controlled trial that tests whether improving the quality of sleep delays cognitive problems, Yaffe said.
One poster presentation did look at sleep apnea treatment, though the study was retrospective rather than prospective. Anne-Cécile Troussière, Université Lille Nord de France, presented her exploratory finding that mild to moderate AD patients with sleep apnea fare better if they use a continuous positive airway pressure (CPAP) device, which treats sleep apnea by applying a mild pressure to keep airways open. The nine patients who refused CPAP treatment declined by three points on the Mini-Mental State Examination (MMSE) over five years, whereas the 28 patients who used CPAP regularly slipped by less than one point. CPAP treatment has previously been reported to slow or improve cognitive decline in AD patients (see Ancoli-Israel et al., 2008, and Cooke et al., 2009).
Another poster supported Yaffe’s suggestion that hypoxia may be to blame for the negative effects of sleep apnea. Emily Clionsky, a private provider from Clionsky Neuro Systems, Inc., Springfield, Massachusetts, found that her memory clinic patients with cognitive complaints, MCI, or dementia have a higher prevalence of hypoxia than previously realized. In her database of 353 patients, 64 percent were hypoxic at some point during the day, either while sitting, walking, or during the night. Previous estimates had put that number at 43 percent (see Reynolds et al., 1985). Clionsky said no good data existed for the prevalence in the healthy elderly population, but that estimates for normal middle-aged people ranged from 2-40 percent.
Taken together, these findings suggest that better sleep benefits the aging brain. In keeping with the theme that one should enjoy everything in moderation, however, some data claim that too much sleep, as well as too little, could be a bad thing. Elizabeth Devore, Brigham and Women’s Hospital, Boston, Massachusetts, analyzed data from the Nurses' Health Study of more than 15,000 women who had been self-reporting their sleep times at mid-life and later in life through periodic questionnaires. From about 2000 until 2006, four separate telephone sessions assessed the women’s cognitive ability. The women were 70 or older at their initial cognitive assessment.
Women who reported the shortest (five hours) and longest (nine hours) sleep durations at both ages had the worst average cognitive scores at the end of the study. Participants in the middle, averaging seven hours of sleep each night, fared the best. In addition, women whose sleep duration changed the most, i.e., by more than two hours per day, between middle and later life, did worse on the cognitive tests than women who slept the same number of hours throughout the study. “In general, these findings indicate that extreme sleep duration or greater changes over time may contribute to cognitive decrements in older adults,” Devore said during a press conference announcing her results. “This could potentially lead to sleep and circadian-based strategies for mitigating cognitive impairment in Alzheimer’s disease.” The accuracy of self-reported data is generally difficult to verify, though Devore did report a correlation of 0.8 between self-report and a six-day sleep diary kept by a small substudy of 260 women. Other scientists cautioned that how long an aging person sleeps may be influenced by many factors.
Claudine Berr, INSERM, Montpellier, France, presented data from the French Three City (3C) Study, which has followed almost 9,300 people in Bordeaux, Dijon, and Montpellier for more than a decade. Surveys at baseline assessed participants’ sleep complaints, asking whether they had excessive daytime sleepiness, were awake during the night or in the early morning, if they had a hard time falling asleep, or if they slept poorly. Follow-up assessments every two or three years looked for cognitive decline, defined as a drop in MMSE score by four points. Reported by about 18 percent of patients, excessive daytime sleepiness alone boosted the risk of cognitive decline by 30 percent. “It is important to recognize and, if possible, to treat sleep problems, particularly excessive daytime sleepiness,” Berr said.—Gwyneth Dickey Zakaib.
This is Part 1 of a two-part series. See also Part 2.
No Available Comments
Like the ebb and flow of ocean tides, Aβ levels in cerebrospinal fluid (CSF) and plasma seem to rise and fall considerably during a single day. One presentation at this year’s Alzheimer’s Association International Conference held 14-19 July in Vancouver showed that age and amyloid status may affect those fluctuations, while another suggested that Aβ accumulation is a culprit in the amplitude of those changes.
“We don’t know for sure what the mechanisms are, but there are really interesting data, including the data presented here, that there is a link between circadian rhythms and Aβ production,” said Francine Grodstein, Brigham and Women's Hospital, Boston, Massachusetts, who chaired a symposium at which some of these findings were presented.
Every healthy adult has a steady, daily rise and fall of CSF Aβ levels (see Bateman et al., 2007). But how do age and amyloid status alter Aβ oscillations? To address this question, Yafei Huang, a member of Randall Bateman’s lab at the Washington University School of Medicine, St. Louis, Missouri, took hourly plasma and cerebrospinal fluid (CSF) samples from three groups of research volunteers over a 36-hour sampling period: amyloid-positive Alzheimer’s disease (AD) patients, age-matched amyloid-negative controls, and young healthy controls with an average age of 36. Each group contained about 10 people. For almost everyone, CSF Aβ42 and Aβ40 levels rose continuously over the total 36-hour sampling period, while plasma levels did not. This rise in CSF levels is a previously observed finding that puzzles researchers. It could be due to a redirection of concentrated CSF Aβ from the brain to a lower area in the spinal cord from the repeated CSF draws, Bateman told ARF. Or it may be from lack of sleep and heightened stress during this study, he said.
Curiously, in this side-by-side comparison of the groups, the rise was pronounced in healthy controls and in older controls without amyloid, but weak for amyloid-positive patients. In fact, for Aβ42, the upward climb was absent altogether. It could be that once plaques are present in the brain, Aβ42 sticks to them rather than crossing into the CSF. This would explain the lack of a CSF Aβ42 rise in the amyloid-positive AD patients, said Huang.
The group also looked at the fluctuations of plasma and CSF Aβ levels over the course of the 36 hours. Canceling out any ascending linear trend, Huang saw cosine curves that fluctuated over 24 hours. The curves' amplitude was highest in young healthy controls, and dropped by about half for both groups of elderly participants. CSF levels were highest around 6 p.m., and plasma levels peaked about midnight, with lows around 12 hours later. “This tells us that it is very important to control for the sampling time when we estimate Aβ levels,” said Huang. “From peak to trough, there can be as much as a 40 percent difference.” Further, given the discordance in Aβ rise and peak times, “we think plasma Aβ levels are not good surrogate markers for CSF levels,” she said.
What causes the circadian rhythm amplitude to shrink in the elderly? Findings from animal studies done in David Holtzman’s lab at WashU, presented by Jee Hoon Roh, link the waning Aβ fluctuation to Aβ buildup. In young APPswe/PS1dE9 mice, both the hippocampus and striatum showed normal ISF Aβ circadian fluctuations. At six months of age, plaques formed in the hippocampus, and circadian variations weakened there. In the striatum, where there were still no plaques at this age, daily fluctuation of ISF Aβ stayed normal. Striatal plaques appeared at nine months, at which time circadian cycles weakened there, too. What’s more, the mice also began to show a disrupted sleep cycle around that age. On the other hand, when the researchers actively immunized mice early on against Aβ, much less plaque accrued and the animals maintained both normal circadian Aβ fluctuation and sleep/wake cycle at nine months. “The accumulation of Aβ in the brain appears to have caused the disruption of the Aβ circadian fluctuation and disturbed sleep/wake cycle, since active immunization to Aβ blocks both abnormalities,” Roh told Alzforum.
Roh presented further evidence from humans that the drop in oscillation results from Aβ deposition rather than aging, as Huang’s study might suggest. He looked at CSF Aβ fluctuation in four middle-aged amyloid-positive and four amyloid-negative presenilin mutation carriers (average age 42.4), as well as four age-matched controls. The CSF Aβ42 levels of healthy controls fluctuated most strongly, followed by levels in amyloid-negative carriers. But amyloid-positive carriers’ CSF Aβ42 oscillation was crimped by half compared to that of negative carriers. Hence, even in middle age, circadian fluctuation of mutation carriers falls with amyloid buildup, Roh reported. If similar abnormalities show up in cognitively normal people and people developing AD pathology, then a dampened Aβ fluctuation may indicate early brain dysfunction, and be useful as an outcome measure in therapeutic interventions, Roh told Alzforum.
Other findings presented at the conference suggest that sleep problems may lead to dementia (see ARF related AAIC story), but those reports use more indirect measures and provide no mechanism for how the connection might come about. Does Roh’s finding hint that the relationship is reversed, i.e., that Aβ42 buildup causes the sleep problems in the first place? Not necessarily, said Roh. “Our data suggest that the sleep-wake cycle and Aβ pathology may have a reciprocal relationship,” he told Alzforum. “The findings reported here suggest that aggregation of Aβ results in abnormalities of the sleep-wake cycle, at least at the beginning.” Previous data from the lab imply that sleep deprivation drives Aβ accumulation as well (see ARF related news story on Kang et al., 2009).
“There are many different pieces here that will hopefully one day come together for a good picture,” said Grodstein. For example, more studies on clock genes, among other experiments, might help unravel the link between Aβ levels and circadian rhythms, she said. “This is a new field, but an incredibly exciting one.”—Gwyneth Dickey Zakaib.
This is Part 2 of a two-part series. See also Part 1.
No Available Comments
On the final day of the Alzheimer’s Association International Conference, attention at the plenary session turned to another common dementia. Frontotemporal dementia (FTD) was the topic of a presentation by Leonard Petrucelli of the Mayo Clinic in Jacksonville, Florida, at the meeting held 14-19 July in Vancouver, Canada. Petrucelli described why sortilin, the receptor for growth factor progranulin, is his “sweetheart of targets” for FTD.
Over the past few years, FTD researchers have linked progranulin, sortilin, and the pathological inclusions containing the protein TDP-43. Nonsense mutations in the progranulin gene cause familial FTD (see ARF related news story on Cruts et al., 2006, and Baker et al., 2006), and TDP-43 shows up in the inclusions that mark many forms of the disease (see ARF related news story on Neumann et al., 2006). Progranulin mutations are generally thought to cause haploinsufficiency of the protein, and that somehow leads to TDP-43’s pathological actions. The manner in which TDP-43 damages neurons is not fully understood, but it evacuates the nucleus and is fragmented, hyperphosphorylated, and aggregated in the cytoplasm.
More recently, scientists have been able to fit sortilin into this picture. A neural receptor, sortilin takes up progranulin and delivers it to the lysosome for dismantling (see ARF related news story on Hu et al., 2010). For its part, TDP-43, an RNA-binding protein, manages both progranulin and sortilin transcripts by keeping progranulin mRNA levels low and regulating sortilin splicing (see ARF related news story on Polymenidou et al., 2011). These manifold connections led Petrucelli to the hypothesis he is now pursuing: If lack of progranulin is the fundamental problem in FTD, then preventing its uptake by sortilin, and subsequent destruction, should support neurons and ameliorate disease.
This hypothesis requires that progranulin be able to nurture neurons via a receptor other than sortilin. A recent paper by Petrucelli and Jennifer Gass indicates that may be the case. Gass analyzed progranulin’s neurotrophic effects in primary hippocampal cultures from mice deficient in either progranulin itself or sortilin. In the cells lacking progranulin, neurite growth and branching were stunted; adding back progranulin solved the problem. To support neurite outgrowth, progranulin required cleavage by elastase into individual granulin segments.
As expected, the sortilin-deficient cultures contained excess extracellular progranulin, since they were unable to degrade it. In these cells, adding more progranulin promoted neurite outgrowth and branching in a dose-dependent manner, similar to its effects in wild-type cultures. This suggests that sortilin is not the receptor for progranulin’s beneficial actions (Gass et al., 2012).
In humans, the reduction of normal progranulin levels may cause neurodegeneration by altering the shape of neurons, the study authors suggested. The team has not yet determined which progranulin receptor nudges neurons toward a growth state. One possibility is tumor necrosis factor (TNF) receptor. Progranulin binds this receptor, and in doing so, blocks inflammatory pathways (Tang et al., 2011). Deficits in progranulin might prompt the cell to follow a detrimental path, the authors speculated, but Petrucelli is far from certain that the TNF receptor is at work in this system.
In Vancouver, Petrucelli also described a detailed study, conducted by Mercedes Prudencio in his lab, of how TDP-43 controls sortilin splicing in human and mouse genes. In human systems, depleting TDP-43 results in more sortilin mRNAs that include exon 17b, compared to cells with wild-type TDP-43 levels where that exon tends to be removed, Prudencio found. In mouse cells, sortilin was more likely to include the exon, even under normal TDP-43 concentrations. This is a major evolutionary difference in sortilin splicing between human and mouse, Petrucelli told Alzforum, and it might explain why FTD mice model only parts of the human condition.
In mouse sortilin, exon 17b, which sits right at the transmembrane domain, makes little difference to the protein’s function. But in human sortilin, this particular exon blocks progranulin uptake, the researchers found. The reason is not that this splice variant cannot interact with progranulin, but that it is secreted instead of settling on the plasma membrane. Petrucelli proposed that this splice variant acts as a “decoy” receptor—soaking up progranulin in the extracellular space and preventing it from binding the unknown receptor progranulin uses to be neurotrophic. Pulling these factors together, Petrucelli’s model suggests that when TDP-43 exits the nucleus, it fails to keep exon 17b out of sortilin, and the secreted sortilin soaks up extracellular progranulin. The result would be that the neuron’s non-sortilin progranulin receptor becomes deprived of the ligand binding it needs to promote cell growth and health.
“Showing a direct link from loss of TDP-43 to a change in sortilin and its potential to uptake progranulin is an important step forward,” wrote Stuart Pickering-Brown of the University of Manchester, U.K., in an e-mail to Alzforum. “This work suggests that blocking the progranulin binding site of sortilin might increase extracellular levels of progranulin,” added Pickering-Brown, who was not involved in the studies.
That is what Petrucelli aims to do. Gass showed that simply adding a half-dozen histidine residues to the carboxyl end of progranulin, which normally binds to sortilin, was sufficient to prevent progranulin endocytosis. Another Petrucelli group member, Chris Lee, used that carboxyl tip as bait in a screen for drugs that would shut out progranulin from the sortilin receptor site. A handful of hits were “quite efficacious” at boosting progranulin levels, Petrucelli reported. Researchers tested one compound in induced pluripotent stem cells derived from a person with progranulin-based FTD, and the drug magnified extracellular progranulin in a dose-dependent manner.—Amber Dance.
No Available Comments
{kind=link}
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.