CONFERENCE COVERAGE SERIES
International Geneva/Springfield Symposium on Advances in Alzheimer Therapy, 2010
Geneva, Switzerland
24 – 27 March 2010
CONFERENCE COVERAGE SERIES
Geneva, Switzerland
24 – 27 March 2010
Last week, scientists from around the world gathered in the Swiss-French border city of Geneva, nestled around the southern tip of handsome Lac Leman. Right from its shores, this old town affords vistas of the Alps’ highest mountain, the Mont Blanc. From 24-27 March 2010, scientists met for the 11th International Geneva/Springfield Symposium on Advances in Alzheimer Therapy to discuss the state of affairs in Alzheimerology with some 90 talks and 60 posters. Your roving Alzforum reporter was able to attend only the first of three days of presentations; hence, impressions of the conference cover merely its N-terminal, or Phase 1, as the case may be. Still, here’s the gist of some of Thursday’s talks for all those who were unable to travel or were in parallel sessions. As always, we invite Geneva-goers to make conference coverage a community project and contribute their impressions.
Why So Few Drugs in AD?
This plaintive title question consumed a full session, as speakers representing stakeholders in AD drug development tried to answer it from their respective perches. A mix of deliberation and some finger pointing provided, if no definitive answer, then themes with which many agreed. Serge Gauthier of McGill University in Montreal started things off with a review of existing AD drugs. He noted that physicians needed better guidelines to decide when to stop prescribing them after their temporary stabilization of symptoms had likely run its course. Gauthier argued for randomized drug trials to start recruiting more homogeneous patient groups as evidenced by biomarkers. He also argued for use of progression milestones as outcome measures, and for more tests of drug combinations.
It’s the Methods, Stupid!
Robert Becker is a co-founding organizer of the original Springfield meetings, which took place in Springfield, Illinois, between 1988 and 1994 at the time Southern Illinois University had an NIA-funded Alzheimer’s Research Center. (The meeting has since moved to various European cities, Montreal, and Hong Kong. Stockholm will host the next one, in May 2012.) Becker has worked in drug development as an academic researcher. He recently founded Aristea Translational Medicine Corporation, which offers error-management consulting to drug developers. In Geneva, Becker argued its message, namely that methodological and execution errors in Alzheimer’s drug development have undermined otherwise respectable drugs. Citing phenserine, flurizan, and metrifonate—a failed drug Becker co-developed—Becker insisted that the drugs themselves were not to blame for the failures but rather errors ranging from trial design and planning to variability introduced by poorly trained raters at far-flung sites. “The methods have really not evaluated the drugs,” Becker said. He, too, noted that biomarkers are the future in AD drug development as a way to reduce human measurement error.
It’s the Drugs, Stupid!
After Becker’s talk, a different wind blew in from the health authorities. Cristina Sampaio is a clinical pharmacologist at the University of Lisbon and serves on the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency. (Formerly known as EMEA, EMA is essentially a decentralized “FDA” of the European Union.) Sampaio noted that while rater and other errors happen in clinical trials, the recent string of Phase 3 failures in AD had multiple, and bigger, reasons. In her view, these came down to the drug companies’ unwillingness to jettison bad drugs early enough. “The regulatory view is that there is no definitive knowledge why so many drugs have failed. I think we have seen well-minded but wrong decisions on the part of companies in taking poor drugs forward,” Sampaio said.
First off, Sampaio noted, scant new drug approvals in AD couldn’t be the result of CHMP’s refusing applications: in the 15 years of its existence, the group has refused but one application for AD, that of metrifonate. Rather, the clinical landscape has changed in that trials are now more difficult to conduct, and fewer approval applications ever reach the authorities. Drug companies are asked to test their candidates on top of existing therapy, and a paradigm shift in the science has moved the focus beyond symptomatic trials toward disease modification, she said. This raises the bar, but has not stopped drug approvals in other chronic progressive diseases, for example, rheumatoid arthritis. There, symptomatic treatments (e.g., NSAIDs) exist side-by-side with disease-modifying ones (e.g., methotrexate or injected biologics).
If it isn’t CHMP’s fault, then whose is it? Target discovery is not to blame, Sampaio said. Plenty of targets exist with reasonable validation and have drugs directed against them in clinical trials. Things mostly go wrong at that late stage. Why? A key shortcoming of current regulatory requirements for trials is the authority’s insistence on clinical endpoints coupled with the lack of formally validated biomarkers. “We recognize that, and we are rushing to validate disease-related biomarkers,” Sampaio said. Trials further suffer from slow recruitment and dropouts that erode their statistical power. That power tends to be weak to begin with, and this is due to variability between patients and between centers, to subjective rating scales that depend on rater training, and to the fact that patients on placebo decline fairly slowly these days. Yes, yes, yes, she recognizes all this, Sampaio said, but added, “I doubt any of the recent failures with disease-modifying trials can be attributed to these types of problems.” Instead, she thinks that bad drugs are kept alive too long before they fail. “People often do not have the courage to kill drugs in Phase 2, because they have already spent too much on them. There is an emotional problem with not going to Phase 3.”
Ideally, intensive screening of potential drugs should weed out the losers sooner, well before Phase 2. Drug-related biomarkers can serve to assess target engagement, and in this way help kill some drugs and help find the right dose for others. Some drug sponsors have not taken this process to heart quite enough, Sampaio said. Nor have they invested sufficient effort into pharmacology and pharmacodynamics before tackling Phase 3, agreed Lon Schneider of the University of Southern California, Los Angeles (see below). This has become a topic of broad consensus among leading researchers. Ron DeMattos of Eli Lilly and Company in Indianapolis, Indiana, as well as researchers from other pharma companies speaking in other sessions in Geneva, detailed their efforts to buttress Phase 3 trials with ample biomarker data to characterize their drug’s behavior from animal research through Phase 2.
From her regulatory perspective, Sampaio described seeing companies take “enormous” risks going to Phase 3 based on flimsy Phase 2 data. In some cases, biomarker-driven proof-of-concept work is hardly done at all, she said; in other cases, when biomarkers flag problems such as CSF data suggesting inadequate brain penetration of tarenflurbil (aka Flurizan, see ARF ICAD story), that data is ignored. She further chided drug sponsors for hiring groups of “magicians” to reinterpret failed Phase 2 data and make them look more palatable for Phase 3. Such data often end up containing more hope than substance, she said. Instead, companies should set strict internal rules for the go/no go decision. “You need those rules. You don’t have to show them to anybody; write them in secret ink and put them in your pockets. But do abide by them,” Sampaio told the drug researchers in the audience, only partly tongue-in-cheek.
When drug sponsors come to CHMP with weak, reanalyzed Phase 2 data and request permission to conduct Phase 3, they should not interpret the committee’s green light as a hint that the agency believes the drug is good, Sampaio emphasized. “If we suspect a drug will fail for lack of efficacy, we will not stop Phase 3 from happening. We only block Phase 3 if we see a safety issue. So our ‘yes’ to conduct Phase 3 does not indicate that we will approve,” Sampaio said. “I really believe the failures happened because the drugs truly do not work and were not killed early enough.”
One force that drives this self-defeating practice is the speed delusion, Sampaio added. Company scientists are under so much pressure to be “first to market” that they skip the essential step of proving that their product crosses the blood-brain barrier, for example, because such a study would slow them down. The way out, she insisted, lies in more extensive use of drug-related biomarkers in preclinical and early clinical stages.
It’s Both, Stupid!
Sampaio’s frank assessment drew endorsement from Lon Schneider, an academic clinical trials expert whose overall take on why drugs appear to have been stalling at Phase 2a could be summed up as: It’s the methods and the drugs.
In listing 28 wannabe AD drugs that flamed out in Phase 2 or 3, Schneider noted that a number of those were actually acetylcholinesterase inhibitors, indicating that even if certain members of a drug class fail (as early immunotherapies and γ-secretase inhibitors did), that does not mean the class can’t be successful later on. One problem in evaluating drugs is that scientists have a poor grasp of where in the long AD process a drug should work best, Schneider said. For example, a drug that helps early on might be ineffective or even toxic later in the process. This issue has come up in trials of estrogen and NSAIDs, as well as anti-amyloids. In Geneva, John Breitner of the University of Washington, Seattle, addressed this issue for the ADAPT trial of the NSAIDs naproxen and celecoxib. For prior coverage of this example, see ARF ICAD story.
AD clinical trials have stayed unchanged in the past 20 years except for minor tweaks around the edges, Schneider said. Some 110 six- to 12-month-long trials and 34 18-month trials in AD by and large have employed the same design and methods because this design enabled approval of the AChE inhibitors. Likewise, patients and their rates of decline on placebo have not changed significantly, either, though it is true that AChE treatment and good medical care can obscure an effective drug if its effect size is very small. All drugs tested so far with statistically significant results have had small effect sizes ranging from about 1.6 to 4.5 points on the ADAS-cog scale for six- or 12-month trials, respectively. Often itself a target of criticism, this scale performs predictably but is relatively imprecise, Schneider said.
The known methodological limitations of trials became a disabling problem because the effect sizes of the candidate drugs tested to date have been très petit. Heterogeneity among patients, ratings, sites, and countries, combined with small effect size, create a situation where a trial is virtually set up to fail if it enrolls fewer than 100 people per group, or if it does not treat for a long time, such as 18 months or longer. Worse, because the trials are underpowered to detect these small effect sizes, play-of-chance rules, meaning that an ineffective drug can come out looking effective.
This same old trial design might still prove serviceable once a drug with a truly strong effect comes along. But for the most part, its limitations make late-stage trials supremely vulnerable to failure, Schneider said, and call for change. Some ideas, besides developing plain better drugs: test them in groups of patients recruited to match the target, for example, anti-amyloid drugs in patients who are known to have brain amyloid; focus on change in individual patients, not groups of patients; use multivariate outcomes that are in line with the expected action of a drug; and bulk up pharmacology.
Hang in There, It Takes Time!
Eric Siemers of Eli Lilly and Company sidestepped the session’s title question. He gave a pharmaceutical perspective on why it takes so long to get to the treatments patients are hoping for. To counter the perception that AD drug development is especially slow, Siemers first noted that while President Nixon’s 1971 declaration of war on cancer has led to more than $100 billion spent by the National Cancer Institute alone, nearly 40 years later therapy has improved, but cancer is far from cured. In Parkinson disease, pharmaceutical drug development to this day revolves around improving dopamine-based symptomatic drugs whose development grew out of a scientific discovery in the 1960s, even though in the interim a handful of PD genes have been identified starting in 1998. No drugs related to protein deposition of α-synuclein, the first PD gene, are in the clinic yet. “It takes a while for industry to shift to other targets,” Siemers said.
Glacial as these timelines seem to someone who has a neurodegenerative disease, by such standards, AD drug discovery would not seem quite such a laggard. Approved in the 1990s to about 2001, current AD drugs followed the scientific discovery of acetylcholine loss by some 20 years. The autosomal-dominant gene discoveries of the 1990s defined a new set of targets against which a “flotilla” of compounds are wending their way through mid- to late-stage clinical trials, Siemers said. One Phase 3 drug, Lilly’s γ-secretase inhibitor semagacestat, first entered the clinic in the year 2000, nine years after the first APP mutation and five years after the first presenilin mutations were reported. In contrast, ApoE has only recently inspired renewed interest in pharma firms. “ApoE is not a highly druggable target,” Siemers said.
It can take a company several years between deciding an initial idea might be druggable and really starting the development process in earnest. “It is difficult for us to shift from dopamine to α-synuclein, or from acetylcholinesterase to Aβ compounds. This is where collaboration and intense discussion can be key,” Siemers told the audience. Moreover, in taking on a new target, companies may use an earlier, inferior compound to explore the concept and keep a better compound just behind the front line. This was in evidence at the Springfield symposium poster session, for example, where scientists from Janssen Pharmaceuticals and Cellzome presented proof of concept for a new γ-secretase modulator in cell culture and in the Tg2576 mouse model, but said they are truly interested only in a less advanced sister compound on which they are not presenting any data. While common in industry, this strategy is expensive, Siemers said.
Constraints outside of the scientific program can delay a drug’s march to market, as well. For example, the number of clinical investigators available to run multicenter trials has been declining for the past decade, Siemers said. Part of the reason might be that companies tend to demand more in the way of procedures and record-keeping from investigators these days, while their payment for a given trial has stayed the same, Siemers said. To enlist enough sites and patients, Phase 3 trials nowadays take place in many different countries all around the world. This provokes some anxiety, Siemers said. While capable people live everywhere in the world, site experience and rater training vary, increasing demand for monitoring. Just like a single national leader arrives at a decision faster than does the United Nations (which incidentally is located across the street from the Geneva International Conference Center, where the conference was held), multinational trials increase the complexity of regulatory oversight, slowing the process down.
Siemers summed up his talk with a wish for more sites, more investigators, more patient involvement, and a simpler regulatory process. On the Holy Grail of finding a Phase 2 strategy that decreases the risk of Phase 3 failure without requiring long treatment periods, he said: “I don’t know what that is. The person who finds it will be able to give plenary lectures for the next five years.”—Gabrielle Strobel.
At the 11th International Geneva/Springfield Symposium on Advances in Alzheimer Therapy, this latter goal—advances in therapy—saw little sprigs of news pop up in talks that otherwise took stock of the current status of immunotherapy as one of the major new approaches in that field. Several speakers started by showing tables listing ongoing immunotherapies that have sprung up since their first demonstration in mice (Schenk et al., 1999). “There is a huge amount of activity on immunotherapy going on in industry,” said Dale Schenk of Elan and Janssen Alzheimer Immunotherapy. Roger Nitsch of the University of Zurich noted that at present, some 10,000 patients are enrolled in more than 40 ongoing trials. The Phase 3 trials—Bapineuzumab from Elan et al., Lilly’s Solanezumab, or Baxter’s Gammagard—are not expected to report results this year. Smaller earlier-stage trials may do so, however. These include one by Genentech, one by GlaxoSmithKline, as well as the ACC-001 active vaccine that represents a second generation to Elan/Wyeth’s AN1792 and is now being developed by Janssen following an agreement between these two companies in late 2009. Several of the current immunotherapy trials are “loaded” with biomarker measurements, said Ronald Black of Pfizer. Solanezumab even advanced to Phase 3 solely on the strength of its safety and biomarker data, without clinical efficacy signals, said Ronald DeMattos of Eli Lilly and company. In fact, so many immunotherapy trials are now being done that companies might be well advised to share experiences on what works and what needs tweaking, said Bengt Winblad of the Karolinska Institute in Huddinge, Sweden.
Generally speaking, immunotherapy trials have begun adopting more innovative designs. They measure CSF Aβ levels and brain amyloid load as well as more distal disease markers such as CSF tau, brain volume by MRI, or, in some cases, brain function by means of FDG PET, though these measures are not usually primary endpoints. The safety record of immunotherapies is a topic of much discussion. For his part, Nitsch said that so far this novel type of therapy appears to be acceptably safe despite some challenges. For active vaccines, pro-inflammatory T cell responses and adjuvant reactions must be avoided. For antibody therapies, vasogenic edemas and microbleeds can occur that appear to be related to dose and to ApoE genotype, and require careful monitoring by MRI. On efficacy, preliminary data suggest removal of brain amyloid and hints of improvement in some responders (Hock et al., 2003; Vellas et al., 2009; Salloway et al., 2009). This latter point is controversial (Holmes et al., 2008) and will remain unsettled until more definitive data come in. Some important differences between active immunization and passive (i.e., antibody) therapies are that in the former, only a minority of patients respond and it cannot be stopped easily, whereas the latter requires more injections and would likely be more expensive, Winblad said.
While new trials are ongoing, scientists are mining the older one for further insights on biomarker data, and that is where some news tidbits were to be had in Geneva. For example, Nitsch previewed a study in press in Brain, in which Alberto Serrano-Pozo and colleagues in Brad Hyman’s laboratory at Massachusetts General Hospital in Charlestown analyzed brain tissue of five more people from the AN1792 trial who had since passed away. Like previous AN1792 autopsy reports, this study documented extensive removal of amyloid from the hippocampus after immunization. One of the five patients had developed meningoencephalitis; this person had some amyloid left in the brain regions Serrano-Pozo examined, and the others had hardly any at all, Nitsch told the audience. Areas of total plaque and of dense core plaques were markedly down. None of these five patients had increased cerebral amyloid angiopathy (CAA) in their brains compared to autopsies of untreated patients, Nitsch added (see also Schenk talk below).
Moreover, Serrano-Pozo et al. noticed that immunotherapy appears to have treated the neuritic dystrophy known in AD. By quantifying how curved (i.e., abnormal) the neurites were in treated compared with untreated patients’ brains, he found that the treated neurites straightened out both in plaque-free areas and near the remaining compact plaques. This would suggest a degree of morphologic recovery of hippocampal neurons, Nitsch said. In addition, the scientists found that immunotherapy appears to have reduced PHF1 immunostaining in the hippocampus of treated patients, particularly those who had not suffered the inflammatory side effect. PHF1 staining reflects hyperphosphorylated tau. In contrast, the scientists saw no change in staining with Alz50, which is thought to reflect a misfolded form of pathologic tau.
Nitsch also spoke about work being done at Neurimmune Therapeutics AG, a start-up biotech company located at the University of Zurich. Scientists there try to develop future therapeutics from naturally occurring antibodies that protect people against AD. Using a trademarked technology called reverse translational medicine (RTM), the Swiss scientists isolate such antibodies from normally aging humans and clone them in vitro. In preclinical experiments, some of these antibodies have lowered the level of pathology in mouse models of neurodegenerative disease, including models of α-synuclein pathology, and delayed disease onset in the SOD1-transgenic model of amyotrophic lateral sclerosis, Nitsch said.
In his talk, Elan’s Dale Schenk noted that because each antibody is slightly different, the 40+ ongoing clinical trials are likely to generate diverging results, as well. Elan is trying to understand its clinical antibodies with crystallography studies (see Basi et al., 2010; ARF related Eibsee story).
Schenk emphasized that as analysis of the first clinical immunotherapy studies continues, its results are beginning to suggest to his mind that mice and humans do show some similarities in their response to immunotherapy, after all. “That's my theme here today,” he said. Previously, mice had been viewed at predicting human outcomes poorly. Specifically, that’s because mice gave no advance warning of the meningoencephalitis that derailed the AN1792 vaccine, and more generally, it’s because clinical failure following murine success stories has become a disheartening theme across neurodegeneration research in recent years (see Alzforum Live Discussion). By now, however, it looks to Schenk as if a growing number of immunotherapy’s established effects in mice—reductions of plaques, of vascular amyloid, of soluble Aβ, of tau pathology, straightening out neurites, behavioral improvement—do seem to occur in humans as well.
Vascular amyloid has become a topic of intense interest, Schenk said. It comes in different forms. Studying amyloid pathology on capillaries of immunized mice, Elan scientist Dora Games and others found that capillary Aβ initially forms during the clearance of plaques and then decreases again over the course of treatment. “This is a temporary phenomenon. We do not know what it means clinically, but we need to factor it in,” Schenk said. Pre-existing vascular amyloid on vessels larger than capillaries corresponds to what in humans is called CAA. Schenk said the 3D6 antibody that formed the basis for Bapineuzumab temporarily increases, but eventually clears, this pathology, noting, “It all depends on when you look.” Microhemorrhages co-localize with vascular Aβ clearance in a subset of blood vessels. In mice, their appearance as measured by hemosiderin staining also depends on dose, and it wanes over time. Finally, Schenk claimed that cerebrovascular amyloid in mice leaves a morphological fingerprint in that the vessel wall is abnormally thicker in some places than in others and that this, too, resolves over time with immunotherapy.
In this context, it is worth noting that Elan/Wyeth in April 2009 dropped the highest dose of Bapineuzumab in its Phase 3 trial of ApoE non-carriers. (ApoE4 carriers, who are assumed to have a higher amyloid burden, only received a single low dose to begin with. See ARF related news story.) Commentators said that Elan took this measure to reduce the risk of MRI flare signals called vasogenic edema; these are thought to be dose-dependent vascular side effects possibly having to do with clearance of Aβ or with inflammation. AD clinicians told this reporter that some of the edemas generate no symptoms, whereas others caused transient dizziness or gait disturbance.
Regarding tau pathology, clinical hints that immunotherapy changes have come from several studies, Schenk said. That AN1792 reduced CSF tau and phospho-tau staining, but not tangles, in autopsy tissue of responders, just like previously in mice, has been reported in published papers (e.g., Gilman et al., 2005), and again by Nitsch in Geneva. Bapineuzumab appears to be doing a similar thing. In its Phase 2 study, CSF levels of phospho-tau decreased as well, though that finding stayed just shy of reaching statistical significance, said Pfizer’s Ronald Black, who presented further clinical data on biomarkers. The hope is that if anti-Aβ immunotherapy indeed reduces tau pathology in CSF and brain, as well as other AD markers, then that would be regarded as evidence for the claim that immunotherapy modifies the underlying disease.
On Aβ reduction itself, autopsy research recently got company when an amyloid imaging biomarker study reported that PiP-PET imaging was able to measure a decrease in living people who had received Bapineuzumab (Rinne et al., 2010; ARF related news story). In Geneva, this finding came up frequently in presentations and hallway discussions. It is considered pharmacodynamic proof that the antibody is bioactive in the brain, though it does not by itself mean the patient will get better.
MRI measurement in immunotherapy trials has been more complicated, Black said. Prior research has established that patients lose volume in the hippocampus and whole brain as their disease progresses (e.g., Barnes et al., 2009); however, in the AN1792 trial, responders unexpectedly lost more brain volume, not less, than non-responders six months after treatment (see ARF related news story). Since then, researchers have tried to understand this finding better, Black said (e.g., Fox et al., 2005). They found, for example, that the areas where the responders’ brains shrank the most were also the areas where there was the most amyloid. “This fits with autopsy data,” Black said. This volume loss stopped one year after treatment. Overall, the researchers suspect that brain areas might temporarily contract because the amyloid is taking water with it as it is clearing out. But the last word is far from spoken, because in the Bapineuzumab Phase 2 trial, MRI data came out as expected. That is, the strongest responders showed less volume loss than did people on placebo. “This was different than in AN1792,” Black said, qualifying that this is preliminary because it came up in post hoc analysis, which can bias the sample.
As for plasma Aβ measurements, this marker went up one day after patients received the Bapineuzumab injection, Black said. Through this kind of research, scientists are learning to distinguish between rapid markers of target engagement, such as a 24-hour plasma signal, and slower, more distal markers hinting at disease modification. In his talk, Ronald DeMattos of Eli Lilly and company also focused on biomarker studies meant to establish separate points: that the brain gets exposed to therapeutic, that the therapeutic engages its target, and that the therapeutic might change disease. Lilly’s research on its Phase 3 antibody Solanezumab has developed different biomarkers for each of these steps, DeMattos said (see prior ARF conference story).
All of these signals are preliminary hints awaiting more definitive clinical data. In the meantime, big unsolved questions remain, Schenk said. Above all, no one yet knows whether reducing Aβ oligomers, plaques, vascular amyloid, and phospho-tau is sufficient to help people clinically with their illness. If so, does it help at the mild to moderate stage, at the early stage, or for prevention? There is a movement afoot to get preclinical studies off the ground. Its supporters, however, have not settled on a specific therapy; they are considering all options including immunotherapy (see ARF API story; see ARF DIAN clinical trials story).
Bengt Winblad of the Karolinska Institute in Huddinge, Sweden, offered the experience of an academic clinician-researcher whose center has already conducted seven different immunotherapy studies for various companies. His site has seen that many participants are highly motivated to contribute to research and help future generations of patients. Their idealism is tested by several practical shortcomings, however. For example, neuropsychiatric testing puts them through a tiring six-hour wringer; shorter visits might improve their compliance. The informed consent form can cause irritation, Winblad noted. Frequently 18 pages long, it serves as much as a legal indemnification package for the sponsor as to inform the patient. Six pages for this form should be enough, Winblad said. Furthermore, many trials require that patients be stable on acetyl cholinesterase inhibitor drugs for six months prior to enrollment. In practice, this may miss a window of opportunity because it means that patients who might be motivated at diagnosis to join a trial lose that interest by the time these drugs have stabilized their symptoms. Some protocols are irksome in requiring an add-on design specifically to donepezil. “We use all the acetyl cholinesterase inhibitor drugs, not just this one,” Winblad said. Participating sites find recruitment difficult when the list of exclusion criteria becomes long and overly restrictive. For example, some trials exclude patients whose blood pressure exceeds 140/80, Winblad said.
Echoing the biomarker theme that pervaded all talks, Winblad recommended that trial designers include baseline labs, CSF and imaging data for each person as a reference for comparison with later findings. He emphasized the importance of having a contact person available around the clock for patients as well as for sites to contact the safety monitoring board. Finally, Winblad urged sponsors to compare notes, especially on practical issues that are independent of their proprietary drug. “As one center running several ongoing clinical trials, it can be dismaying to see that different sponsors make similar planning errors.”—Gabrielle Strobel.
This story ends with a glimpse of the newest kid on the block. Called 18F AZD4694, this radioligand does not yet have a mellifluous name (à la “flor…” “flute…”) that rolls easily off the tongue. It follows AstraZeneca’s equally numerical 11C ligand AZD2184, which is identical in structure to PIB save for a carbon-nitrogen substitution to reduce white matter binding. That 11C ligand is published and its performance looks strong as a research tool (Nyberg et al., 2009; Johnson et al., 2009); however, its 11C radio nucleotide limits its distribution and wide clinical applicability as much as PIB’s.
The 18F ligand made its first showing at the AD/PD meeting in the Czech capital of Prague last March, where AstraZeneca’s Samuel Svensson presented preclinical data (see ARF comment). Ever since then, rumor had it that it might actually be the best of the 18F bunch, but no clinical data were available up until now. 18F AZD4694 was absent from the HAI program, but at the 11th International Geneva/Springfield Symposium on Advances in Alzheimer Therapy last March in Geneva, Switzerland, that data finally came out. AstraZeneca’s Zsolt Cselényi presented on a poster the first results of the Swedish pharma company’s ongoing evaluation in people. It featured collaborative work with scientists at the Karolinska Institute in Stockholm, including Maria Eriksdotter Jönhagen and Christer Halldin. Also at Springfield, Eric Reiman of the Banner Alzheimer’s Institute in Phoenix, Arizona, presented his take on the Swedish data as well as the Banner group’s own investigation of this new ligand. Unlike AstraZeneca’s 11C compound, the 18F one is structurally quite different from PIB; in chemical parlance, PIB is phenyl benzothiazole, whereas AZD4694 is piridinyl benzofuran.
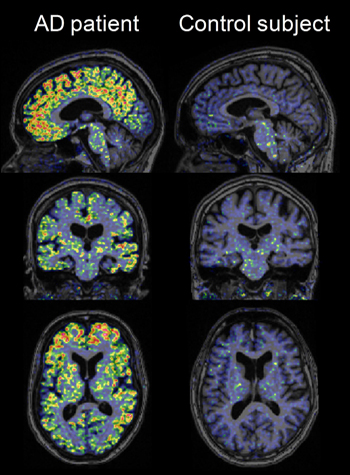
High-resolution research tomograph PET images obtained using [18F]AZD4694 in a probable AD patient and a cognitively normal elderly control subject, overlaid on structural MRI. Image credit: Zsolt Cselényi, AstraZeneca
In the Swedish study, 10 people with mild to moderate AD in their fifties to seventies, plus six age-matched controls, underwent a PET scan after injection with this ligand; three AD patients and four controls repeated the same procedure later that day to check if the ligand performed consistently in what’s called test-retest. At Banner, Reiman and colleagues Dan Bandy and Kewei Chen tested AZD4694 in four people with very mild AD, five cognitively normal older adults, and one 23-year-old control.
Both presentations noted that AZD4694 rapidly sweeps into the brain and out again. Reversible binding peaked around 27 minutes after injection. This suggests that for routine clinical purposes, patients could get away with spending shorter periods of time—five to 20 minutes—in the scanner than is customary with the other ligands. The Swedish poster made this point, and Reiman confirmed it based on a different analysis of the Swedish data. AD patients can become anxious and disoriented in the scanner. The fast kinetics also imply that sites can start imaging some 25 minutes after the volunteer has received the injection, a shorter-than-usual wait.
A standard data representation in PET imaging called the time-activity plot showed a separation in how long the ligand remained in the cortex versus the cerebellum of an AD patient; in a normal control these curves overlapped. This, among other data, is considered evidence of specific binding. The compound did so in brain regions of interest in AD, including the prefrontal cortex, anterior and posterior cingulate cortex, and other areas. On the poster, plots of each person’s uptake values in the Swedish study, both by SUVR or DVR, for each of these AD regions of interest suggested showed no overlap between AD and controls in this small initial group of 16 people. With more use, this separation will likely blur somewhat because a fraction of cognitively normal older people are known to have brain amyloid, and occasionally, ostensible AD patients turn out to have been misdiagnosed. Indeed, among the Banner volunteers, this overlap already appeared. Reiman showed a plot of mean cortical SUVR, not split by region, but the point is it showed that the ApoE4 carriers among the cognitively controls retained AZD4694, one of them near the AD range and one in this range.
In Geneva, both Reiman and Cselényi showed that the ligand appears to bind white matter less than the field has grown accustomed to seeing from the other known 18F ligands. This enhances contrast between the specific labeling of amyloid plaques and the rest of the brain, making scans easier to read. White matter binding at times may complicate reading the scans a bit, Reiman said. (AZD4694 does bind non-specifically in the brain stem, however.) Indeed, images of AZD4694 in people with and without AD looked crisp to the unaided eye. The Swedish images delineated gyri and sulci of the cortical folds with a clarity not typically seen on amyloid PET scans. That wasn’t the doing of AZD4694 alone; these images were taken on a high-resolution research tomograph (HRRT) at the Karolinska Institute. Few neuroimaging labs worldwide have this cutting-edge machine available to them.
For his part, Reiman stopped short of endorsing AZD4694 just yet. Its low white matter binding in particular would appear to make it the ligand of choice for imaging small amounts of amyloid at the preclinical stage of AD, in ApoE4 carriers (see ARF related API story), and in presymptomatic carriers of eFAD mutations (see ARF related DIAN story). In the latter, this ligand might afford a fresh look at puzzling findings, whereby PIB scans differ significantly from scans of late-onset AD in showing strong striatal amyloid and relatively less cortical amyloid until later into the disease.
But Reiman emphasized that a clear answer to the question of where each ligand’s respective strengths and weaknesses lie could only come from a direct comparison of different ligands in the same AD and control participants and using the same imaging system, reference region, and image analysis techniques. “This new tracer has significant promise, and I am excited about it. But hold your horses until we see head-to-head comparisons,” Reiman said.—Gabrielle Strobel.
This is Part 5 of a six-part series. View a PDF of Part 5. See also Part 1, Part 2, Part 3, Part 4, Part 6. View PDF of the entire series.
No Available Comments
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.