CONFERENCE COVERAGE SERIES
International Winter Meeting: Growth and Death in the Nervous System
St. Moritz, Switzerland
24 – 28 March 2004
CONFERENCE COVERAGE SERIES
St. Moritz, Switzerland
24 – 28 March 2004
In the waning days of March, researchers from around the world met in St. Moritz to send off the cold season with three days of science and skiing amid the glorious scenery of the Engadine Alps, in one of Switzerland’s most beautiful valleys. At 200 participants, it was the largest conference since the Swiss Society of Neuropathology began hosting this series 20 years ago. Titled “Growth and Death in the Nervous System,” the meeting was expertly organized by Adriano Aguzzi, Markus Glatzel, and their colleagues at University Hospital of Zurich. It featured a blend of science on prion diseases, Alzheimer’s, Parkinson’s, gliomas, as well as fundamental processes that cut across individual diseases, such as inflammation, proteasome function, and protein folding. The talks and discussions brought out intriguing parallels between research into prion diseases and Alzheimer’s, for example, an emerging focus on oligomers in prion research, or the struggle to come to grips with the problem of misfolding of PrP, Aβ, and α-synuclein . This summary is updated with more recent publications on some of the covered topics.
Part 1
Prion Diseases: Few Answers, Many Questions
Charles Weissmann of the MRC Prion Unit, London, set the stage by reviewing current knowledge—and the most glaring knowledge gaps—of transmissible spongiform encephalopathies. In this group of diseases, neurodegeneration, ataxia, and dementia usually sets in after age 60. Once overt, disease takes a rapid course of about one year, although the incubation period may last many years. (Symptomatic Alzheimer’s progresses more slowly, though this disease, too, begins with a silent pathogenic phase of a decade or more.)
Human prion diseases include Creutzfeldt-Jakob disease, which can be sporadic or familial, the familial Gerstmann-Straussler-Scheinker syndrome and fatal familial insomnia. More notorious have been the acquired cases, either through tainted growth hormone injections, transplants, cannibalism rituals (kuru) or eating BSE-infected animal products (variant CJD). Prion diseases most obviously differ from Alzheimer’s in prevalence: they are all rare. Moreover, they have the added complexity of being infectious, with patients showing no immune reaction to the infectious agent. Alzheimer’s has never been considered a transmissible disease, and in practice still isn’t, though Mathias Jucker challenged this tenet for the sake of a fascinating argument.
Genetics has been less generous to prion researchers than to their colleagues in Alzheimer’s. Though more than 20 rare PrP mutations are known in families with familial CJD, no other causative, modifying, or risk-factor genes have since turned up. In Alzheimer’s, 4 genes are definitively known, and while the search for further ones has been a slog, linkage or association studies have at least produced many additional candidates that are awaiting independent confirmation (or invalidation) in separate patient samples or by newer methods. Genetic mosaicism is a potential factor in both prion diseases and AD, (see Beck et al., 2004). For more on neurogenetics, see John Hardy’s talk.
It took 30 years to purify the scrapie agent (scrapie is a prion disease of sheep), until Stanley Prusiner’s group at UCSF pulled it out of infected brain, Weissmann said. Its main component is PrPsc, a pathogenic and infectious form of the normal cellular glycoprotein PrPc. When years of searching for scrapie-specific nucleic acids came up empty, the field generally (though not completely) accepted Prusiner’s Nobel Prize-winning contention that the infectious unit contains only protein.
A major open question in prion disease is simply: What causes disease? Two hypotheses advanced for this question have not panned out, Weissmann said. One implicates a slow-acting virus, the other (the “virino” hypothesis) points to a yet-to-be discovered small oligonucleotide. The leading contender these days postulates that an abnormal version of the normally expressed host protein catalyzes the conversion of further PrPc into more abnormal forms. PrP knockout mice made in Weissmann’s lab by Hansruedi Bueler are healthy and normal. Surprisingly, the mice stay healthy when challenged with intracerebral inoculation of PrPsc, showing clearly that the cellular form of PrP must be present for prions to multiply, spread through the organism, and cause disease. This insight has raised important questions about the precise protein-protein interaction between PrPc and PrPsc.
Another nagging question is: What, precisely, is the infectious agent? “It’s the thing that causes prion disease,” quipped Aguzzi, adding that facetious as it may seem, this is as good an answer as the field can offer. Scientists can convert PrPc to PrPsc in a cell-free system, however, no one has been able to show that this protein is then fully infectious. “To this day, we cannot identify the infectious agent and study its structure,” said Weissmann. Similarly, perhaps, Mathias Jucker reported that endogenous but not synthetic Ab was able to seed amyloid deposition in mouse brain.
When mature PrPc converts to PrPsc, it becomes partially protease-resistant. This old finding has become a staple of prion studies, but its pathogenic significance, if any, remains unclear, Weissmann said. Meanwhile, several investigators have cloned and expressed PrPc and PrPsc. Nobel laureate Kurt Wuethrich and his colleagues at the ETH in Zurich have solved the NMR solution structures of human, mouse, and bovine PrPc expressed in E. coli, and Wuethrich described their structural similarities and differences in St. Moritz. However, no one has yet solved the atomic structure of PrPsc. Like Aβ and α-synuclein , PrPc folds only partially, and contact with PrPsc turns the partially folded form into a conformation that is rich in β-sheet. Which other factors or environmental conditions stimulate the conversion of protease-sensitive, α-helical PrPc into insoluble, protease-resistant, β-sheeted PrPSc is the million-dollar question in prion research, many scientists agreed. Curiously, RNA reenters the picture here, as some labs have suggested that host RNAs might stimulate this crucial step (see Adler et al., 2003, Deleault et al, 2003).
Two hypothetical models exist for how PrPc might convert, Weissmann said. One is a stoichiometric notion that one PrPsc refolds one PrPc; the other is a “seeding model.” The latter posits that PrPsc would slowly form a crystallization seed while the equilibrium is strongly on the PrPc side, but then additional PrPsc would bind rapidly to this seed and form large aggregates. Peter Lansbury formulated this model for AD and scrapie (Jarrett and Lansbury, 1993; since then Lary Walker (Walker et al., 2002 and Mathias Jucker, among others, have expanded upon it but the precise process of protein misfolding remains unclear for both PrP and Aβ.
Both Weissmann and Aguzzi emphasized that another of the puzzling questions in prion research concerns the existence of strains. Scientists can characterize and propagate different strains from animals. How can this be, and what does the word strain even mean when the prion is a protein of one given sequence? Prusiner suggested that different conformations might account for this, as was later shown. New insight on this issue came out of a separate line of research dealing with prion-like elements that occur in yeast. This at first seems confusing because yeast prions are made of proteins other than PrP. Certain yeast proteins can spontaneously convert to a different conformation, and this change gets propagated onto further proteins, which then lose their function. One protein that converts in this way is the translation stopper sup35p. Weissmann quoted a recent paper from Jonathan Weissman (no relation) and colleagues at UCSF showing that different temperature conditions cause sup35p to convert into physically different structures (it forms amyloid fibers at 37 degrees) (Tanaka et al., 2004). The study provides an example for how different conformations of a protein give rise to distinct, stable prion strains driven by an environmental condition. Incidentally, prions occur not just in yeast and mammals. Prion-like properties have recently been reported in Aplysia synapses (see ARF related news story).
Finally, as in AD, no robust biomarker exists for prion diseases; the presence of PrPsc is the best surrogate marker available to date, Weissmann said. On this topic, Glatzel described Swiss and English cases of human CJD who deposit PrPSc in their skeletal muscles. His lab is investigating increasingly sensitive detection methods of intramuscular PrPsc to evaluate their potential as diagnostic tools (see also Kovacs et al., 2004).
In a related finding, French researchers yesterday reported in Nature Medicine that they detected PrPSc in muscle of scrapie-infected sheep. They spotted the first traces months before the sheep showed the first symptoms of disease (Andreoletti et al., 2004). While this work overlaps with Glatzel’s regarding the potential for developing future diagnostics, it may also stoke public fears about eating lamb and mutton. Andreoletti et al report that the infectivity of the muscle PrP was 5,000 times lower than that of brain PrPsc.
Bruce Chesebro, of NIH’s Rocky Mountain Laboratories in Hamilton, Montana, presented an extended analysis of how pathology develops after PrP infection in mouse models created in his lab. Species barriers limit this line of investigation, but it turns out that transgenic mice expressing hamster PrP develop disease when infected with hamster PrPsc. Chesebro showed images of tufts and plaques of prion deposits in mouse hippocampus. At first glance they resemble Ab amyloid deposits, but at higher magnification they clearly look extracellular as well as intracellular in neurons, astrocytes, and around blood vessels. Electron microscopy showed deposits near the plasma membrane and in lysosomes. These mice also have marked degeneration of dendrites and neurons. Chesebro compared PrP accumulation in astrocytes and neurons across different mouse strains that express transgenic PrP either in astrocytes, neurons, or both. This led him to suggest that whatever pathogenic changes occur in astrocytes lead to secondary, indirect damage of the neurons. As in AD, variability in transgene expression levels and the compressed time span of the pathogenic process confound the extrapolation of such data to humans. Chesebro’s lab is currently analyzing the role of microglia in prion pathogenesis. He also presented study of prion deposition and neurodegeneration in the mouse retina as a model organ that is more accessible than brain.—Gabrielle Strobel.
No Available Further Reading
Anthony Williamson, of the Scripps Research Institute, La Jolla, California, first noted that to make further inroads into the disease, the field sorely needed an atomic structure of PrPsc. He agreed with Weissmann and Aguzzi that one of most pressing issues is to reveal the neurotoxic molecule, and that a way to get there was to identify what auxiliary molecules nudge the conversion of PrP. Williamson cited grafting studies showing that PrPsc by itself is not neurotoxic but needs a host accomplice and, more recently, a study where genetic interruption of neuronal PrPc expression during an ongoing CNS prion infection prevented neuronal loss (see ARF related news story).
But besides PrPc’s interaction with PrPsc, it may also have a direct role in neurotoxicity, Williamson said. This is why he thinks uncovering PrPc’s normal function is important. Perhaps PrPc delivers a signal in vivo? To pursue this idea, the Scripps researchers developed an anti-PrPc antibody injection experiment. Results suggest that when PrP interacts with the antibody on the cell surface, it dimerizes and in this form then delivers an apoptotic signal to the neuron (see ARF related news story). In prion-infected brain, oligomeric PrP might have a similar effect as did the antibodies in this experiment, Williamson speculated. With regard to possible relevance to AD, formation of APP dimers or multimers is not heavily studied but has received some attention as a potential neurotoxic mechanism (see Scheuermann et al., 2001, Lu et al., 2003). Williamson noted that his data calls for caution in developing therapeutic antibodies for prion diseases.
In work that dovetails with Williamson’s, Corinne Lasmezas suggested that she observes in vitro the fatal consequences of dimerizing PrP in the membrane that Williamson’s group has modeled in vivo. Lasmezas works at the Neurovirology Service of the Atomic Energy Commission in Fontenay aux Roses, France. She presented new data on the possible role of oligomeric PrP species in the pathogenic mechanism. Her approach is part of a new avenue in prion research and appears to re-trace more established efforts in AD research to get an experimental handle on Aβ oligomers and their effects. Tau and α-synuclein oligomers are also under study. Put simply, the reasoning behind this work is that on their way to form large aggregates, proteins must pass through small oligomeric states. Might they be toxic? Lasmezas described different PrP peptides that lodge firmly in lipid bilayers. An inspiration to her current work came from Detlev Riesner at Heinrich-Heine Universitaet in Duesseldorf, Germany, who had described intermediate species of recombinant hamster PrP: a dimeric α-helical one and a tetra- or oligomeric β-pleated sheet structure (Jansen et al., 2001).
With distinct oligomeric species available for study (resulting either from a transient beta-sheeted monomer or from a covalently linked dimer intermediate), Lasmezas asked whether they might be toxic to neurons. To test this, she added the oligomers to cultured primary cortical neurons and assayed how the cells functioned and survived. Both types of oligomer were highly neurotoxic, Lasmezas reported, regardless of whether the cultured neurons expressed endogenous PrP. Certain PrP-binding antibodies protected against this toxicity, as did heparan-sulfate mimetics, which interfere with PrP interactions at the cell surface. Interestingly, the oligomers made of covalent dimers were not infectious, suggesting that separate mechanisms are responsible for infectivity and neuronal death. Lasmezas speculated that PrP oligomers have exposed hydrophobic side-chains that are normally covered. This would make them more prone to slide into the membrane bilayer and interact with membrane components or receptors in such as way as to signal apoptosis.
Questions from the audience echoed concerns frequently raised in the AD field: where exactly are these oligomers? Are they physiologically relevant? How can one visualize them and make them amenable to study? Researchers in AD still struggle with the technical difficulty of demonstrating the relevance of oligomers in vivo (but see ARF related conference story ), and there is no proof in humans. Even so, this line of research has won a growing number of converts over the last decade and led to a shift in the amyloid hypothesis.
In his presentation, Aguzzi described experiments aimed at coming to grips with the molecular mechanism of prion replication. His group picked up on the idea that a PrP—PrPsc heterodimer might form at some point, designed such a soluble dimer, and expressed it in transgenic mice. Crossbreeding showed that this dimer, dubbed PrP-Fc2, did not restore the vulnerability to infection of PrP knockout mice. Yet when crossed back into wild-type mice, the dimer blocked PrPsc accumulation following PrPsc infection in brain or spleen. In effect, when interacting—somehow—with normal PrPc, the soluble dimer slowed pathogenesis by interfering with deposition of the protein-resistant PrPsc, Aguzzi said. Immunoprecipitation experiments suggested the soluble dimer relocates and slides into lipid rafts only after the mouse has been infected, Aguzzi added. His group is currently using this dimer construct to try to identify any ancillary factors that some scientists still suspect are present when PrPc converts to PrPsc.
Aguzzi then reviewed current knowledge on the question of how prions get to the brain, a topic also studied by speaker Moira Bruce at the Institute for Animal Health in Edinburgh, U.K. In brief, prions enter the brain through the peripheral nervous system. They usually reach the spleen within days of infection but can take months or even years before entering the brain. They get in by way of the sympathetic innervation of the spleen. Cutting this organ’s innervation in mice delays neuroinvasion, whereas mice with excessive splenic innervation will show prions in their brain more quickly. Several labs focus their effort on defining the connection between the autonomic nerve endings and cells in peripheral organs such as the spleen. However, as with the nature of the prion, the exact point of this neuro-immune border crossing remains elusive, Aguzzi said.
What is the physiological function of PrP? It probably has one, otherwise kuru epidemics that may have raged through prehistoric human populations (Mead et al., 2003) would have selected for PrP-negative people. As in AD research, where the function for APP is nebulous and for Aβ even more so, prion researchers don’t have an answer to this question, either. And as in AD, researchers disagree on whether it even is a crucial question. Some speakers, including Williamson, think yes. Lasmezas said PrP might play a role in synaptic signaling and neuron survival related to its ability to regulate copper levels in the synaptic cleft. PrP interacts with heat shock proteins, other membrane receptors, and heparan sulfates from the extracellular matrix. Aguzzi, however, thinks not. “We should find out what that function is, but we really do not know that it will have anything to do with the pathogenesis. Once we know the function, it may prove irrelevant,” he said.
On this issue, a tantalizing mystery revolves around the Doppel (from “Doppelgaenger”) gene, which lies just downstream of PrP. The protein can interact with PrP and may, in fact, antagonize it (see also LeBlanc comment). Structurally and biochemically, the doppel protein resembles a form of PrP with its amino-terminal domain clipped away, and both can be neurotoxic in vivo. Normally, though, the brain expresses only minute amounts of doppel. To study this strange protein-protein relationship further, Aguzzi’s group knocked out Doppel and, when the knockout mice proved unable to breed, discovered that doppel is required for sperm to form properly (Behrens et al., 2002). While this is no answer to the question of PrP function, it suggests that perhaps a place to look for this answer is not the brain, but the testes, Aguzzi said.—Gabrielle Strobel.
No Available Comments
In the study of genetics, bursts of excitement alternate with periods of lulls. John Hardy, of NIH in Bethesda, took advantage of a present lull (i.e., no new genes on offer this time) to integrate current knowledge in neurodegeneration genetics into a bigger picture. The book is closed on autosomal-dominant versions of most neurodegenerative diseases, as most genes are found, Hardy said. The burning question today is what causes sporadic disease. “On that, I have an almost embarrassingly simple message,” he said. Put as a campaign slogan it would be: "It’s quantity, stupid." Protein deposition is a mass action process, and when the concentration of a given protein rises to a critical point, it becomes insoluble and aggregates. Then it interferes with cellular pathways in myriad ways that will vary from disease to disease. Even so, the fundamental problem is a rising tide of aggregation-prone proteins, and at least part of the reason for this, Hardy proposes, lies in genetic variability of their expression levels (see also Singleton et al., 2004).
The writing was on the wall as early as 35 years ago, when it became clear that people with Down’s syndrome develop AD (Olson and Shaw, 1969), Hardy said. In his famous paper describing the amyloid protein, George Glenner presciently proposed that dementia in Down’s is simply caused by overexpression (Glenner & Wong, 1984).
In Alzheimer’s, the primary deposited protein is a fragment of a gene product, and the mutations found to cause rare Mendelian AD (in APP and the presenilins) increase its production. To find out what’s going on in the vast majority of AD cases, Hardy’s and other labs have screened the genome in sibling pairs of AD disease. They found that AD siblings share their given APP gene variant more than they should by chance. While Hardy has not identified particular high-risk alleles, he speculates they will prove to be the high-expressing ones. It’s not yet clear whether variability in presenilin expression also contributes. In a separate but related vein, recent work has indicated that BACE activity may increase with age.
Diseases marked by tau-only pathology include FTDP 17 as a genetic form plus sporadic versions of essentially the same condition, called supranuclear palsy or corticobasal degeneration. Genetic variability in either tau expression or tau splicing determines risk for these diseases, Hardy suspects, but at present his data can’t differentiate between the two.
Hardy and others noticed that human populations have two tau haplotypes, which differ in intron size. Each is inherited as a block. Curiously, this haplotype is a bizarre structure that, at 2 megabases long, is fifty times larger than haplotype blocks typically are, Hardy said. His group is trying to find out why no recombination would have occurred over such a long stretch of DNA. Homozygosity for the H1 tau haplotype is a risk factor for sporadic tauopathies, as 95 percent of patients have two copies of it, while only 60 percent of the general population do. The H2 haplotype occurs only in people of European ancestry, and both H1 and H2 are more similar to the chimpanzee tau haplotype than to each other. Half in jest, Hardy tossed out an idea that is far-fetched but worth a closer look. Could the H2 haplotype be a genetic legacy of Neanderthals? “A question in anthropology is: Did the Neanderthals kill or breed with contemporary early humans? If history is any guide, they probably did the former with the men and the latter with the women,” Hardy said.
In prion diseases, the primary depositing protein is PrPsc. The first family with a prion mutation was described in the late 80s. In sporadic disease, John Collinge showed that homozygosity at the PrP locus is a predisposing factor, whereas being heterozygous for a common polymorphism confers some resistance. The human PrP locus is unusual in its high degree of heterozygosity; indeed the HLA locus of antigen-presenting genes is one of few other examples known in the human genome, Hardy said. Last year, Collinge suggested that epidemics of kuru-like disease decimated prehistoric populations and in the process created a selective pressure for PrP heterozygosity in human evolution (Mead et al., 2004). In a similar way, evolution is thought to have selected for heterozygosity for the sickle cell gene because it protects against malaria. And yet, while PrP homozygosity increases risk for prion disease, genetic variability at the PrP locus is an even more important factor. The simplest explanation there, too, is that variations in expression levels account for the differences, Hardy said.
Lastly, Hardy discussed diseases with Lewy body pathology, that is Parkinson’s disease (PD) and dementia with Lewy bodies (DLB). Here, too, mutations in α-synuclein can cause familial disease, and genetic variability in the promoter of the normal gene affects one’s risk of developing sporadic PD (Farrer et al., 2001), possibly through higher expression (Chiba-Falek & Nussbaum, 2001). The clearest evidence for the effect of genetic variability on disease risk comes from a family whose early onset PD traces back to a founder triplication of the α-synuclein gene in the 19th century. This family has no mutation, but members who inherit the affected chromosome end up with 4 copies of the α-synuclein gene instead of the normal 2. “This is the Down’s of Parkinson’s,” Hardy said (see ARF related news story). In Huntington’s disease, too, early indications hint that high-expressing promoters contribute to risk of sporadic disease. Last but not least, genetic variability at the SOD locus should be studied with regard to risk for developing ALS, Hardy added.
Mathias Jucker, who last year joined the new Hertie-Institute for Clinical Brain Research in Tuebingen, Germany, agreed with Hardy that increasing concentrations of proteins prone to misfolding and aggregation are the fundamental commonality between these diseases. He studies the question: once protein concentration has reached a critical concentration, how, then, does aggregation begin?
First, however, Jucker asked where brain amyloid comes from. Prior work had suggested that it enters the brain from the periphery, or that local microglia, smooth muscle cells, or neurons produce it. Jucker favors the latter. He pointed, for example, to analysis of the rare hereditary disease cerebral hemorrhage with amyloidosis-Dutch type, which shows that the amyloid derives from neurons even though most of the deposits build up around blood vessels (see also ARF Live Discussion). To address this question experimentally, his group recently generated a new strain of mice that expresses human APP only in central neurons, not in smooth muscle cells.
This is how Jucker suggested amyloid forms: Neurons release Aβ from the cell body or synapses, interstitial fluid transports it over long distances, and then it enters peri-arterial drainage and gets cleared into blood and the lymphatic system, see also Roy Weller’s presentation. A concentration-dependent, stochastic seeding process along the interstitial pathway explains how deposits form in the vessel wall and in the spaces between neurons. Weller noted that seeding may begin in the vessel wall, and that this may then block the perivascular flow and drainage, resulting in accumulation of Aβ upstream in the parenchyma, particularly while neurons keep releasing Aβ.
To test the idea further, Jucker picked up the concept of seeded polymerization developed by Peter Lansbury, and shown in vivo by Lary Walker. Then at Pfizer, Walker injected AD brain extract into the neocortex of young Tg2576 APP transgenic mice too young to develop plaques on their own. Then he documented the formation of massive amyloid pathology near the injection site and its spread from there (Walker et al., 2002). Jucker extended this work with single injections of diluted brain extract from old APP23 transgenic mice into the brain of their young counterparts. These experiments confirmed Walker’s in that massive amyloidosis formed around the injection site in a dose- and time-dependent way, in which seeding was the rate-limiting step. This happened only in mice that express human Aβ, not in wild type.
Much like prion researchers scratch their heads over precisely what their infectious agent really is, Jucker also asked: What is the seed? Intriguingly, none of many concoctions of synthetic Aβ the scientists injected was able to seed aggregation at the low concentrations that brain extract did. And yet, the seed must be a form of Aβ, because mixing the brain extract with anti-Aβ antibodies prior to injection, or injecting antibody into previously seeded mice both prevented seeding. This suggests that some mysterious feature of endogenous, human Aβ is necessary to induce aggregation. “It may be a folding issue,” Jucker said, “where we cannot reproduce the pathogenic conformation of the native Aβ with synthesis. Or it may be an additional factor that makes Aβ refold in such a way that it becomes a seed, such as a proteoglycan or a chaperone that is present in vivo but not in vitro. Like in prion research, this comes down to structural biology.”
Finally, to jolt awake an audience that was fading at 9:30 pm after 12 talks and a poster session, Jucker asked whether his and Walker’s experiments make AD a transmissible disease, and Aβ an infectious agent. Granted, delivering the brain extract orally did not seed amyloid, and intraperitoneal injection experiments are still ongoing, Jucker said. Even so, the echoes with prion diseases are eerie. There, too, debate revolves around what makes PrPc assume the conformation of PrPsc, and whether this happens in an interaction between PrPc and PrPsc or together with an external factor X? Like in prion disease, amyloid pathology arises from an interaction between a normal and a mutant form of the protein at hand. PrP knockout mice cannot be infected with PrPsc, and even people with familial AD are heterozygous, meaning they have both normal and mutant APP, Jucker said.
Whatever the answer to this tangle of questions, Jucker believes that in sporadic AD, seeding occurs as a stochastic process once Aβ levels rise due to slight increases in production or clearance problems of various kinds. Together, his work also adds indirect support to therapeutic efforts targeted at early stage amyloid formation, Jucker said.
Intersecting well with Jucker’s work is the hypothesis presented by Roy Weller of University of Southampton School of Medicine, England. Weller proposes that sporadic AD develops as the aging brain gradually loses its ability to clear Aβ along perivascular fluid drainage pathways.
Several mechanisms act in parallel to rid the brain of excess Aβ: Carrier proteins mediate its uptake into blood (see ARF Live Discussion), microglia and astrocytes ingest it, and enzymes such as neprilysin, IDE and others degrade Aβ locally. In fact, Hasan Mojaheri of the University of Zurich, presented experiments extending Takaomi Saido’s original work with neprilysin. Mojaheri told the audience that mice upregulate neprilysin in response to challenge with Aβ, but that the enzyme can only degrade non-aggregated forms of the peptide, and neprilysin induction makes a difference only in young, pre-symptomatic mice.
But back to Weller, who studies a fourth clearance mechanism, namely transport along blood vessel walls. Vascular amyloid usually accompanies AD, and the anatomical pattern of amyloid within artery and capillary walls in the brain suggests that Aβ deposition may begin with its entrapment in the narrow drainage pathways there, Weller hypothesizes. To visualize these pathways, the researchers injected soluble dextran tracers roughly the size of Aβ into adult mouse brain and then co-localized them with laminin to the basement membrane of capillary and artery walls. The resulting pattern of deposition and drainage models Aβ drainage, Weller said.
Cerebrovascular disease induces an age-related congestion of interstitial flow of Ab because arteriosclerosis reduces the amplitude of arterial pulsations, which generate the putative motive force for the perivascular drainage of Ab, Weller said. Evidence for this is that thrombotic occlusion of cortical arteries results in accumulation of Ab in vessel walls upstream of the thrombus, he added. Weller and James Nicoll contributed a synopsis of this hypothesis to a recent on vascular factors in AD, see Alzforum discussion. See also Weller and Nicoll, 2003 and Preston et al., 2003).
How to Study a Folding Protein? Grab It, Watch It.
If indeed the misfolding of an accumulating protein marks a crucible for pathogenesis, how is one to study it? On this question, Harald Janovjak stood in for principal investigator Daniel Mueller, both at the Max-Planck-Institute of Molecular Cell Biology and Genetics in Dresden, Germany, to highlight what the developing field of atomic force microscopy could do for neurodegeneration researchers. The Mueller lab has spent the last few years working out protein imaging and structure analysis methods with the atomic force microscope (AFM), and some may just be ready for prime time.
This desktop instrument offers two general advantages. First, that it allows one to image proteins in relatively native conditions, such as in physiologic solutions or embedded in artificial membranes, and do so at a handsome resolution of up to 0.5 nm. Secondly, it’s possible to physically grab a single folded protein, pull it taut, release it, and measure the tiny, sequential forces with which it resists the unfolding and then snaps back into shape. These force traces then enable the scientist to make inferences about structural characteristics of the protein.
Having a crystal or NMR solution structure of the protein at hand is helpful but not necessary, Janovjak said. In fact, the AFM can predict the outline of a protein’s secondary structure, and such predictions have been confirmed by atomic structures elucidated later on.
Janovjak illustrated some new AFM capabilities. For example, he showed images of gap junctions that visualized individual connexon units. Moreover, the scientists imaged a conformational change as the gap junctions closed in response to an increase in calcium (Muller et al., 2002).
Furthermore, the AFM allows the researcher to image intramolecular forces. For example, consider pull-and-release experiments on bacteriorhodopsin, a complex, well-studied intramembrane protein. The tip of the AFM stylus is attached to a flexible cantilever. Mueller and colleagues retracted the cantilever, measured the force-extension spectra, and correlated these spectra to the extraction of that single protein. This analysis told them that pairs of two transmembrane helices unfold pairwise and in this way act as one structural unit. The German scientists also detected particular secondary structure elements, and studied differences in how they unfold and then refold into their configuration when released (Muller et al., 2002). Finally, Janovjak showed examples of how tinkering with the experimental conditions—temperature, pH, ion concentration—can affect the folding of a given protein (Janovjak et al., 2003).
This ability to modify fairly native conditions experimentally is but one reason why the AFM might lend itself to the study of Alzheimer’s and prion diseases, Janovjak suggested. Another is that many of the proteins of interest are located in membranes. In AD, the role of cholesterol and its metabolites, as well as their possible effect on APP processing, has become a research priority. What’s more, APP proteolysis may well occur in lipid rafts, and their composition is thought to change with age. Could one, for example, insert APP into lipid membranes of different composition and see how such environmental changes affect its folding? The Muller lab invites suggestions for research collaborations in the area of neurodegenerative diseases at (Mueller@mpi-cbg.de).
Retro Traffic Reigns in Protein Renegades?
Weller and Jucker study events occurring after Aβ has left the neurons. But some protein misfolding and oligomerization may well begin inside neurons, see for example Takahashi et al., 2004). How does the neuron try to defuse this ticking time bomb of misfolding and aggregating proteins? This question got attention from Wiep Scheper at the Academic Medical Center in Amsterdam, the Netherlands. Scheper studies Rab6A, a small GTPase that functions in the trafficking of proteins through the ER/Golgi membrane network. Previous work on APP trafficking and presenilin had pointed Scheper’s attention to Rab6, and in St. Moritz she reported data on more direct experiments testing its possible role in AD. Working with Frank Baas and colleagues, Scheper found that, in AD post-mortem brains, Rab6A is upregulated and predominantly expressed in pyramidal neurons of the temporal cortex and hippocampus.
Five years ago, German researchers had found that Rab6 travels along a then-newly discovered retrograde transport route from the Golgi back to the ER (White et al., 1999, Girod et al., 1999). Normally, misfolded proteins are caught in the act in the ER, expelled through the Sec61 channel and fed to the proteasome for degradation. Yet some proteins elude this quality control, and make off into the Golgi on their way to the plasma membrane. Scheper suspects that this Rab6-dependent retro route serves to return such escapees to the ER. She cited prior indications that Rab6 might function in quality control (Luo and Gallwitz, 2003, including of the PrP protein (Beranger et al., 2002). Scheper studies Rab6A in relation to the ER chaperone BiP and reported a close correlation between the two in human brain, possibly because Rab6 induces BiP. This work is ongoing, but Scheper suggested that aging, mutations, or proteasome inhibition may overwhelm the cell’s capacity to retrieve and degrade misfolded proteins. —Gabrielle Strobel
No Available Comments
Unlike prion researchers, their colleagues in Alzheimer’s disease have a rich trove of current and potential future drug targets into which they can sink their collective investigative teeth. It is, of course, the APP processing problem. Unlike PrP, APP is chopped up in a complex series of enzyme reactions, some of them right in the middle of a lipid bilayer, which involves three different proteases and generates the Aβ peptide as well as other fragments. The hope is that understanding this processing—which occurs in most cells of the body but over the course of decades leads to amyloid buildup only in the brain—will yield new treatment approaches beyond the ones that are currently in preclinical and clinical development.
In St. Moritz, Christian Haass of Ludwig-Maximilians University in Munich, Germany, took the audience on a tour, punctuated with plays on famous jazz compositions, of how far the field has come since the demonstration that presenilin is indeed the catalytic subunit of γ-secretase (Wolfe et al., 1999). Since then, it has become clear that the proteins nicastrin, aph-1, and PEN-2 join presenilin to form a γ-secretase complex. The cutting edge of research therefore has moved to ask, for example, just how the four arrange themselves into the unusual kind of enzyme that is an intramembraneous aspartyl protease. Another current question is whether this complex of four is complete or involves further, unknown players, especially in human neurons (Haass, 2004).
All four proteins in this quartet are equally important, and they keep tabs on each other in a process Haass dubbed “coordinated regulation.” Once this issue is fully resolved, it will probably be clear that all players interact with one another, Haass said. For now, his lab has established a sequence of protein-protein contacts and assembly that goes roughly like this: Immature nicastrin binds to aph1 and then folds itself properly. Next, presenilin binds nicastrin, and it does so by dipping its normally cytosolic C-terminus into the membrane and binding it to a transmembrane spot on nicastrin. Lastly, PEN-2 joins the trio. At this point presenilin is still inactive; it occurs in a pro-form until it gets cut through its long cytoplasmic loop and then comes together as an active heterodimer. Prior data from Takeshi Iwatsubo’s group suggested that PEN-2 might mediate this activating internal cut. Having recently tested this idea, Haass and his colleagues now suspect that the self-cleavage happens as an independent autoproteolytic event, and that PEN-2 then comes in to stabilize the emerging presenilin heterodimer (see Prokop et al., 2004). Where does all this happen? The γ-secretase quartet assembles itself in early areas of the membrane continuum that is the endoplasmic reticulum, Haass added. The complex does not move up to the plasma membrane to meet APP until it is fully put together.
The second question Haass addressed concerns whether the γ-secretase quartet is complete or needs further players. In parallel with other laboratories, his group tried to reconstitute γ-secretase activity outside of neurons, and finally pulled it off in yeast, which has no endogenous γ-secretase activity (Edbauer et al., 2003). “This is a simple model but it took five years to get it to work,” Haass said. Using this model, the researchers recovered Aβ38, 40, and 42. They also found that this yeast γ-secretase cleaves twice, once to release Aβ and then a second time to release the AICD fragment that is thought to translocate to the nucleus and regulate gene expression there. These two findings validate that the artificial yeast system recapitulates key characteristics of the neuronal γ-secretase, Haass said. All this does not imply that human γ-secretase acts as a foursome, Haass added, saying that many other proteins may well participate and modulate this reaction in vivo.
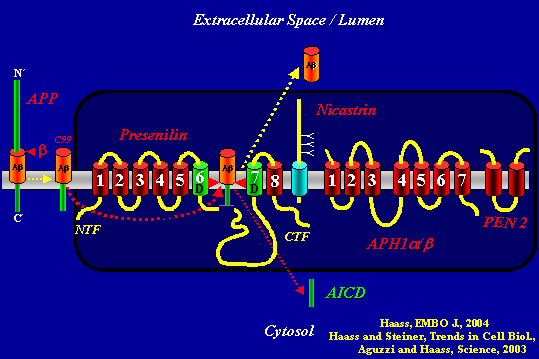
This slide summarizes the fate of APP when it is not cut by α-secretase. Dark box: APH1 binds Nicastrin, which binds the C-terminus of Presenilin. Next, PEN 2 binds to this trio and stabilizes the presenilin heterodimer that comes out of presenilin’s autoproteolysis of its long cytosolic loop. All this occurs in the ER. The complex then moves to the plasma membrane, where it meets APP. After b-secretase generates the C99 fragment from full-length APP, γ-secretase cuts C99 twice. First it releases Aβ into the extracellular space (or the lumen of an intracellular membrane compartment) and then AICD into the cytosol, from where it can signal to the nucleus.
Bart De Strooper of KU Leuven, Belgium, continued by saying that the yeast model was a technical feat, in part because it showed definitively the minimum requirements needed to get γ-secretase going. At the same time, human presenilin complexes likely assemble in a variety of forms. This real-life variability perhaps holds out hope for future, more specific γ-secretase drugs, especially if making a small dent in its activity were sufficient. “γ-secretase is a complicated activity and we are only beginning to understand it,” de Strooper said. He and Haass agreed that none of the current crop of inhibitors can separate APP from notch (see ARF related news story). Indeed, even partial PS knockouts de Strooper’s lab created to model γ-secretase inhibition more like a drug would do it and less like a complete deletion came down with autoimmune problems that one can expect if T cell maturation goes awry.
The γ-secretase variations—different complexes forming in different tissue, for example—may be necessary because the biological role of this enzyme is to help the degradation of a range of membrane proteins by cutting them right in their transmembrane sections. For example, mice harbor 3 Aph1 genes, and initial data indicate that conditional knockouts of Aph1a, b, and c generate different phenotypes. In a broader program, De Strooper’s lab is selectively deleting parts of the complex and monitoring the effect on APP and other substrates.
De Strooper’s postdoc Diana Dominguez presented initial data on their BACE1/2 double knockout mice. BACE is a favored drug target in part because single BACE knockout mice develop and age normally (see Luo et al., 2003). In practice, BACE may prove more recalcitrant, de Strooper said. For one, the protein is difficult to inhibit, though this is a technical hurdle the pharmaceutical industry may overcome in time. For another, new data suggests that inhibiting BACE might cause side effects, too. BACE 1 and 2 are the only members of this group of aspartyl proteases, and some data indicates that 2 might act to restrain 1. To get a better sense of how BACE1 and 2 interact, and of their role in vivo, Dominguez created mouse lines that lack BACE1, BACE2, or both.
The double knockout mice are fertile but, puzzlingly, half the litters die by three weeks of age. The scientists don’t yet know why, but early suspicion has fallen on germs in the mouse facility. Perhaps these double-knockouts are particularly sensitive to infection.
Finally, de Strooper outlined new research on a physiological function of APP. Studies in fruit flies indicate that it may help the brain recover from traumatic injury. Stay tuned for more on this story later.—Gabrielle Strobel
No Available Comments
Two scientists updated the audience on the continuing workup of patients in Elan’s ill-fated phase II trial of the AN-1792 vaccine. James Nicoll recounted what he got to see firsthand from his perch as a research and diagnostic pathologist at the University of Southampton’s General Hospital in England, when one of the eight English trial patients died a year after she’d fallen sick with probable meningoencephalitis (see ARF related news story). Since then, three more patients have died, and their brains are under investigation in Spain (Ferrer et al., 2004), California, and Cardiff, the latter two still unpublished. Initial results of these three additional cases show they had identical features to the Southampton case, Nicoll said. This suggests that Aβ immunotherapy can indeed remove some Aβ from the brain, but with complications that need resolving if this is ever to be a viable therapy.
In summary, the pathology profile that emerges from looks like this: Most strikingly, wide swaths of parietal and temporal cortex were devoid of plaques. Instead there were isolated patches of amyloid, and quantification showed an overall reduction. Aβ-reactive microglia were prominent, as well as amyloid around the blood vessels. This was all as predicted by prior animal models, as was the finding that dystrophic neurites straightened out, which has been described in mice, as well (Lombardo et al., 2003; for picture of neurites see ARF related news story. Yet there were unexpected findings, too. The pathologists were surprised to see that T lymphocytes and macrophages had infiltrated the brain, that there was evidence of white matter pathology, and that neurofibrillary tangles and neuropil threads remained stubbornly in place. On this last point, Isidro Ferrer’s study of the Spanish patient noted some indications that tau phosphorylation, and by implication further tangle formation, appeared to have slowed somewhat, which one could hope if indeed Aβ spurs tangle formation (Goetz et al., 2001).
What, then, caused this serious side effect? Nicoll suggested these possible explanations: First, microglial activation could have spun out of control in the heat of Aβ removal. Second, the solubilization of Aβ brought on by the antibody could have overburdened clearance mechanisms, effectively increasing Aβ flux, concentration, and then deposition in the perivascular pathway enough to cause the leucoencephalopathy. Third, T lymphocytes infiltrating across meningeal vessels could be the culprits. This last notion gets support from an independent study of seven people with CAA-related inflammation who had rapid cognitive decline and white matter disease, but got better when given immunosuppressive therapy, just like the Elan patients did (Eng et al., 2004). "These seven cases described by Steven Greenberg look remarkably similar to what we see," Nicoll said.
How about the immunized people who still live? The public has not yet seen an analysis of the patients from any location of this multicenter trial with the exception of the Swiss cohort, which is being followed by Roger Nitsch’s group at the University of Zurich.
Nitsch prefaced his lecture, titled “Can Antibodies Bring Back Memories?” by noting some of the prior data that convinced him of Aβ’s value as a therapeutic target. Standing out were Brian Bacskai’s and Brad Hyman’s multi-photon images showing the disappearance of plaques in mouse brain (Bacskai et al., 2002). “It was a shock to us who believe it takes 10 years for a plaque to develop to see it disappear within three days after a single dose of antibody,” Nitsch said.
This website has covered the Nitsch group’s initial report of the 30 Swiss patients extensively (Hock et al., 2003 ), as well as a prior update Nitsch offered at last year’s Society for Neuroscience conference in New Orleans (see ARF conference report ). Therefore this summary will simply add some details not previously mentioned. Nitsch noted that two years after the patients received their last vaccine injection, they still maintain high levels of IgG and IgM antibodies. These antibodies do not cross-react with APP or its derivatives, but specifically label amyloid deposits in brain tissue of AD and CAA patients. The antibodies are present in serum and in the cerebrospinal fluid. Nitsch said he suspected there are being transported across the blood-brain barrier by receptors.
Nitsch also offered an explanation for a data point that has drawn criticism. The “non-responder” patients who did not produce antibodies declined by six points on the MMSE scale in the first year of assessment, an unusually steep drop by some measures. However, the trial patients were in their 4th year of taking cholinesterase inhibitor drugs, and after the initial effect of these drugs wears off, patients are known to decline more quickly. Since the 19 “responders” all improved, they are unlikely to be outliers, Nitsch added.
The cases of subacute aseptic meningoencephalitis that arose in 18 of the 298 patients vaccinated worldwide are clearly related to the vaccine, Nitsch confirmed. Yet antibodies probably had little to do with it, as their levels did not correlate with it, and some of the people who developed it but recovered are among the highest responders clinically.
On the mechanism of Aβ clearance, Nitsch noted that Nicoll’s observation of activated microglia and amyloid-free patches of brain hinted that Fc-mediated clearance was at work. To try to test the peripheral sink hypothesis (see, e.g., DeMattos et al., 2002), Zurich researchers Christoph Hock and Uwe Konietzko analyzed blood samples collected from the patients every month, but they were unable to find changes of Aβ42 levels in plasma. Neither did they see significant changes in CSF. Future directions being explored for better immunotherapy approaches include passive vaccination, more careful epitope selection, and anti-inflammatory strategies, Nitsch concluded.
Privately, several scientists at the conference voiced concern that all patients of the halted Elan trial may not be followed as carefully as the Zurich cohort, raising concerns that a valuable learning opportunity might be lost to the field.
Microglia: Good or Bad? Conditional K.O. Offers an Answer
Quite a separate question in immunotherapy, and more broadly in neurodegeneration and neuroinflammation, revolves around the role of microglia. To this day, these ‘macrophages of the brain’ remain shrouded in mystery, and scientists trying to lift this shroud variously come across contradictory results. In general, research finds that normally quiescent microglia become activated in most brain diseases, and that they begin spewing oxygen radicals, nitric oxide, cytokines and chemokines in the process. Yet is this activation beneficial (as in removing amyloid) or detrimental (as in killing neurons)? At what point does one tilt over into the other? In short, precise mechanisms of how microglia function in the pathogenic process remain unclear. To probe the role of microglia in vivo, Frank Heppner at Zurich’s Institute for Neuropathology created a clever transgenic mouse line that essentially allows him to selectively silence microglia. Working with Aguzzi and others, Heppner started out with a promoter specific to monocytes/macrophage (microglia are of hematopoietic origin.) Behind it he spliced a suicide gene that makes the otherwise inactive pro-drug ganciclovir toxic to proliferating cells. Heppner’s twist to this widely used system lies in transplanting wild-type bone marrow into these transgenic so that the chimeras had normal, ganciclovir-insensitive hematopoietic cells in the periphery but transgenic, ganciclovir-sensitive microglia. “Ganciclovir only hits the brain macrophages,” Heppner said.
To confirm that the transgene worked as expected, Heppner cut the facial nerve of these mice, an operation that activates microglia without affecting the blood-brain-barrier, in other words avoids confounding infiltration of peripheral macrophages. Unlike control microglia, which proliferated, the transgenic microglia did not respond to the severed nerve at all, Heppner said.
Next, Heppner used his model to define the role of microglia in multiple sclerosis, a demyelinating neuroinflammatory and, in its later stages, neurodegenerative disease known to involve both T cell and microglial activation. The question was who is doing what in the pathogenesis, and are the two responses independent of each other? When the scientists injected the MOG peptide antigen to induced an established mouse version of multiple sclerosis called EAE, they noticed that the ensuing T cell response occurred in the transgenic mice as expected, showing that microglia are not necessary for the peripheral immune system to mount its autoimmune response. However, the mice with the impotent microglia barely got sick. They had a much milder disease with later onset and they recovered, unlike normal mice given the same injection. Inflammatory infiltration of the spinal cord and cerebellum were also muted in the transgenic mice. This suggests that taking microglia out of commission represses the clinical course of multiple sclerosis, and establishes these cells as valid drug targets, Heppner said. Precisely what did the trick remains to be shown, however; candidate mechanisms include that the microglia were unable to present antigens (see ARF related news story) or the absence of the noxious substances that activated microglia usually release.
On the broader front of mouse genetics, Klaus Rajewsky, Harvard Medical School, introduced the audience to new techniques of generating mutant models. In collaboration with Rudolph Jaenisch at MIT, Rajewski is working out methods that could do away with the cross-breeding of single-mutant strains by which so-called compound mutant strains are currently being generated. Cross-breeding is laborious, expensive, and introduces confounds stemming from strain backgrounds. Instead, the scientists are devising methods to grow mice from embryonic stem cells that already contain all the desired mutations.
Anders Nykjaer presented his data, recently published in Nature, on nerve growth factor signaling (Nykjaer et al., 2004). Working with Claus Petersen at Aarhus University in Denmark, Nykjaer analyzed how a new receptor family called sortilins might influence the signaling of NGF. Part of the reason why NGF signaling is complicated lies in its ability to promote both cell survival and cell death, and to do so via different receptor types. Simply put, NGF generally signals survival through TrkA receptors, while its immature form, pro-NGF, signals cell death via the receptor P75NTR. Both functions have an important role in shaping the developing nervous system; in neurodegenerative diseases, NGF could potentially be a treatment were it not for the difficulties of delivering it to needy neurons in a targeted way. Earlier basic research had indicated that additional receptors likely participate in transmitting NGF’s death signal. Nykjaer and colleagues suspected that sortilin, which had been described a few years ago but had few functions assigned to it yet, might be the one. In St. Moritz, Nykjaer reviewed data showing that sortilin binds pro-NGF tightly in its pro-domain whereas, in the same complex, p75NTR binds to the ligand’s mature NGF domain. This appears to fit with new crystallographic images of NGF-p75NTR binding (see ARF related news story). Sortilin antagonists can protect against cell death induced by pro-NGF. By contrast, sortilin does not interact with TrkA, Taken together, this suggests that sortilin essentially functions as a switch that can “sort” life from death.
All told, this conference drew on some of the liveliest neurodegeneration science, particularly from Europe. As with every good meeting, this Alzforum summary cannot cover nearly all of the 43 talks and 51 posters but instead highlights selected presentations and common motifs. As always, the writer invites conference participants to fill the gaps, and indeed all readers to comment on any of the points raised.—Gabrielle Strobel.
No Available Comments
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.