Earlier this month in Washington, D.C., 95 scientists from 23 companies, 19 academic institutions and two regulatory agencies met with funders, advocates and patients and caregivers. The buzz was all about learning from the mistakes and setbacks of drug development in Alzheimer’s disease and getting a collective act together while the FTD field is still young. On what did the group agree? Basic science and longitudinal human studies are advancing apace, but what the fields needs most urgently now to launch more and good trials is a toolbox of biomarkers to subtype FTD disorders and measure target engagement. For their part, the regulators want creative, rigorous science that tries to couple biomarker change to meaningful outcomes, but assured the scientists that no disease is too rare for them to be keenly interested and approve drugs for it. Read Gabrielle Strobel’s series.
Drug Trials in Frontotemporal Dementia: Can Field Push Forward Together?
On March 31 and April 1, 95 scientists from 23 companies, 19 academic institutions, as well as public and private funding and related organizations gathered in Washington, D.C., for the Frontotemporal Dementia Study Group’s fourth workshop. While the city’s cherry blossoms bloomed riotously outside, the scientists huddled in a hotel basement room with advocates, family members, and people with FTD. They took stock of how far the field has come in its quest to understand this heterogeneous set of diseases, and what tools they most urgently need to develop medications to treat or prevent them. Importantly, the group got an opportunity to quiz three leading regulatory scientists for advice on how to develop drugs for them (see Part 3 of this series). Disorders across the FTD spectrum encompass behavioral variant FTD, three different forms of primary progressive aphasia, progressive supranuclear palsy, corticobasal degeneration, and FTD with motor neuron disease. Together they afflict 50,000 to 60,000 people and their families in the United States. No approved treatments exist.
Overall, eight hours of meetings imparted four lessons:
The basic science on FTDs is creating unprecedented momentum.
The setbacks of Alzheimer’s drug development hold valuable lessons.
Biomarkers are beginning to emerge, but much more work is needed.
The Food and Drug Administration and European Medicines Agency urge rigorous science that links biomarker change to meaningful clinical outcomes.
Emphasis on openness and data sharing resonated throughout. The vision of a shared infrastructure platform for Phase 1/2 FTD trials was briefly floated, but while the regulators in the room welcomed the idea, pharmaceutical companies appeared not quite ready to embrace this deeper level of collaboration.
“FTD is a young field. We are encountering a friendly and open world, with lots of advice from Alzheimer’s and Parkinson’s,” said Susan Dickinson of the Association for Frontotemporal Dementia in Radnor, Pennsylvania. “You have a fantastic opportunity here. We at the agency are enthusiastic about greater attention to these horrible diseases,” William Dunn of the Food and Drug Administration told the assembled audience. “It’s amazing how fast the FTD field has gone from description of syndromes to molecular pathophysiology, discovering neuroimaging metrics and biomarkers. This happened much faster than the decades it took the Alzheimer’s field to get to where FTD is now,” said Geoffrey Kerchner of Genentech in South San Francisco, California, which is developing a therapeutic tau antibody and a tau PET tracer. “Let’s acknowledge the importance of this meeting. These kinds of partnerships are critical to achieve our goals,” said Brad Dickerson, who runs a large FTD clinic at Massachusetts General Hospital. See below for a detailed summary.
The AFTD and the National Institute of Neurologic Disorders and Stroke jointly sponsored the conference. Dickinson noted that despite broad international consensus on diagnostic criteria for FTDs, most physicians still do not have these diseases on their list of potential diagnoses for middle-aged people who present with marital strife, disordered behavior, or failing language. That said, attention in research and industry has picked up tremendously in the past few years. For its part, AFTD alone has increased research funding from $35,000 in 2005 to $3 million in 2016, and a handful of other philanthropies have sprung up to support specific FTD subtypes. Together these groups have assembled a community of patients and gene carriers who want to participate in trials. They also want a say in decision-making along the way. “Patients and their families are here today, as well. They are ready to do their part in the work,” Dickinson said, adding that every board member at AFTD cares, or has cared, for a relative with FTD.
The organizers did not come empty-handed. Chief among the new funding opportunities they brought to the meeting was the $5 million FTD Biomarkers Initiative. This invites proposals to discover new, or develop existing, biomarkers for any disease across the FTLD spectrum. Company scientists are welcome to apply, too, said Nadine Tatton of AFTD, which co-funds drug discovery grants with the Alzheimer’s Drug Discovery Foundation (ADDF). For its part, the NINDS recently announced a funding opportunity for a multi-institutional center without walls to identify and validate molecular mechanisms that contribute to tau pathogenesis and associated neurodegeneration in FTD. This U54 grant addresses the top basic science recommendation from the NIH ADRD FTD working group. The winning proposal will use existing infrastructures, and feature ample data and resource-sharing as well as collaboration with non-governmental organizations and philanthropy, said NINDS’ Margaret Sutherland. In addition, NINDS announced PAR 16-122, a grant focused on parkinsonism that could support biomarker and clinical data collection for progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), two forms of FTD that lie on the parkinsonian end of the FTLD spectrum. At the NINDS Alzheimer’s and Related Diseases Summit, held at NIH earlier in the week, Ron Petersen of the Mayo Clinic, Rochester, Minnesota, estimated that about $161 million of the $991 million overall federal ADRD research budget would be spent on the related diseases, which include FTD (see Apr 2016 conference news).
The Challenge: A Varied, Complex Biology
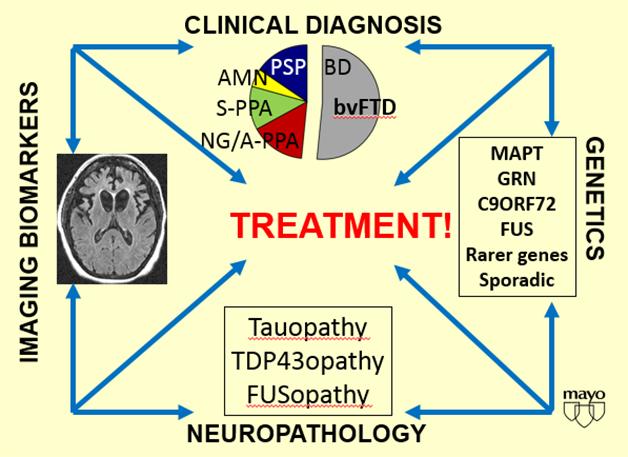
The heterogeneity of the frontotemporal dementia spectrum makes drug development a great challenge, said David Knopman of the Mayo Clinic in Rochester. Its two main pathologic lesions—tauopathy and TDP-43 proteinopathy—each occur as several different subtypes, and debate continues about what form of the two respective proteins constitutes the toxic species. Mutations in three main genes—tau, progranulin, C9ORF72—plus several minor ones cause frontotemporal dementia. The genes map somewhat consistently to particular pathologies, though a large proportion of FTD appears sporadic.
Frontotemporal dementias all feature neurodegeneration in the front and side of the brain (left); beyond that, their clinical expression, genetic origin, and underlying protein pathologies are highly heterogeneous. [Courtesy of David Knopman.]
Perhaps most puzzling for trialists: Most FTDs show no clear link between their clinical picture and a given protein pathology. This means that when a patient comes to an FTD clinic, even experienced diagnosticians do not know if they are dealing with tau or TDP-43, or indeed FUS, yet another molecular pathology in FTLD. It also means that a therapy trial cohort enrolled by way of clinical criteria and MRI, but without a biomarker that ascertains an underlying molecular pathology, might constitute a mix of people with tau, TDP-43, or FUS pathology. This may be workable for neuroprotective or other drugs that target common mechanisms of neurodegeneration, but for tau-based drugs, it could dilute the number of people in the cohort who are likeliest to respond to the therapy. For example, the bvFTD cohort of the current TRx0237 Phase 3 study was enrolled in this way.
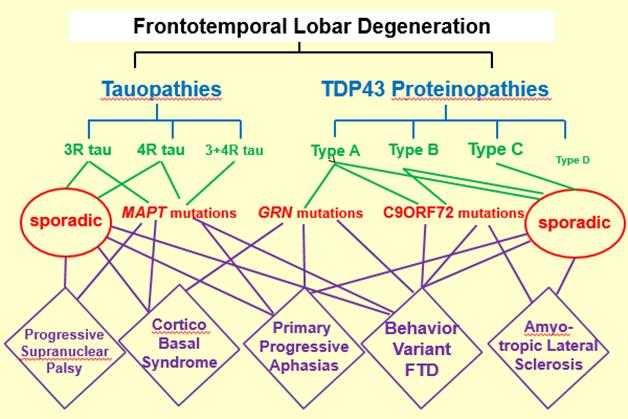
bvFTD is indeed the enfant terrible of the lot. It is the most common form of FTD, but a full 15 etiologies have been described to underlie its clinical presentation, said Bill Seeley from the University of California, San Francisco. A more typical example is primary progressive aphasia. This language disease can be sporadic or caused by progranulin or tau mutations, and feature either TDP-43 or tau pathology. An exception is PSP, a sporadic disease that almost always features tau pathology. This clear clinic-pathological relationship, combined with the disease’s rapid progression and the existence of a serviceable rating scale, has made PSP patients sought-after participants for a growing number of small trials of candidate drugs targeting tau.
How do you run trials in this? Most clinical types of FTD can have one of several underlying pathologies. Mutations in a given gene lead to different clinical diseases. [Courtesy of David Knopman and Dennis Dickson.]
This heterogeneity, compounded by the small number of available patients, means that successful therapy evaluation will require biomarkers for a diagnosis and for target engagement for each molecular subtype of FTD.
The Status: Work in Models to Find Molecular Mechanisms
On the tau side of the FTLD spectrum, the heterogeneity is apparent in distinct cellular pathologies and rates of progression between one person’s disease and another’s. It may stem in part from the manifold post-translational modifications tau undergoes and in part from tau’s ability to form strains. Strains are structural variants of tau that cause a particular form of tau pathology. They can be isolated from diseased human brain and faithfully passaged between cells and mice. “Strains are the building blocks of tauopathy,” said Marc Diamond of UT Southwestern Medical Center in Dallas.
The TDP-43 proteinopathies in particular have experienced a surge of interest. Of the 1,837 papers published in PubMed since the TARDBP gene was cloned in 1995, 1,825 appeared since Manuela Neumann and Tetsuaki Arai implicated TDP-43 in neurodegeneration (Oct 2006 news); 1,000 have appeared in the last four years alone. (Not all these papers are on TDP-43 alone, some merely mention it.)
This nuclear RNA-binding protein regulates its own production and, for poorly understood reasons, it ends up in the cytoplasm where it accumulates in a variety of structures. “We must, for therapeutic purposes, understand what causes this mislocalization,” Leonard Petrucelli of Mayo Clinic in Jacksonville, Florida, told the assembled FTSG audience. Nuclear export and import has become a prominent theme in understanding TDP-43 aggregation.
The protein’s C-terminus hosts mutations causing ALS and some cases of FTLD. Functionally, this end mediates formation of stress granules and cytoplasmic aggregates. The protein’s N-terminus is thought to regulate multiple different aspects of the lives of hundreds of different target RNAs, from pre-mRNA splicing to microRNA processing, non-coding and long non-coding RNA metabolism, as well as mRNA transport, stability, and translation (for open-access review, see Ratti and Buratti, 2016).
Researchers have generated a host of TDP-43 mouse models to study whether gain-of-function versus loss-of-function underlie toxicity (see Alzforum Research Models Database). Biomarkers are farther behind. No PET tracer is yet in sight, though the Swiss biotech company AC Immune and Biogen on April 18 announced a partnership to develop one (see company press release). Current CSF or blood assays work poorly, in part because peripheral cells produce more TDP-43 than the brain. This makes it difficult to define how much of a given concentration in a fluid sample represents brain pathology, said Kaj Blennow of the University of Gothenburg, Sweden.
Like TDP-43, C9ORF72 has taken the FTD research community by storm. It has generated 684 PubMed entries since Rosa Rademakers and Brian Traynor discovered, in 2011, hexanucleotide repeat expansion in this gene. On April 18 at the American Academy of Neurology’s annual meeting in Vancouver, British Columbia, these two neurogeneticists received the 2016 Potamkin Prize for Research in Pick’s, Alzheimer’s, and Related Diseases (see Apr 2016 news).
C9ORF72 expansions are the most common genetic cause of FTD, ALS, or ALS-FTD. Mutation carriers have aggregated TDP-43 inclusions in their central nervous system, and the C9 expansion has been linked to mislocalization and aggregation of TDP-43. “How that happens is a key question in the field these days,” said Petrucelli. More broadly, the abiding research question for C9ORF72, just like for TDP-43, is whether the DNA expansion is toxic by way of a loss of the gene’s normal function, or gain of toxic function. The cellular function of the C9ORF72 protein is poorly understood, but it is implicated in endosomal/autophagy-related membrane trafficking and thought to play a role in innate immunity. Even so, researchers do not currently favor haploinsufficiency as the main pathogenic mechanism of C9ORF72 diseases. A toxic gain of function could result when RNA transcripts of the C9 repeats aggregate into foci that entrap cellular RNA-binding proteins. It could also arise from defects in transport between nuclear and cytoplasmic factors, or from translation of the DNA repeat expansions into aggregating dipeptide repeat proteins that could damage proteasome degradation or cause ER stress, Petrucelli told the audience.
Clinically, dipeptide repeat pathology has been placed earlier in the C9ORF72 disease process than TDP-43 inclusions, but attempts at staging disease remain in their infancy. Three mouse models, but no fluid or imaging biomarkers, are available for study (see Alzforum Research Models). For an open-access review of C9ORF72, see Todd and Petrucelli, 2016.
The Status: Work in Humans to Chart Evolution of FTD
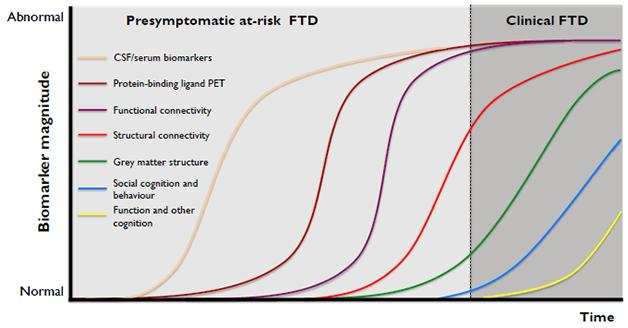
Parallel to research in mice and cell-based models, initiatives are underway on both sides of the Atlantic to characterize the natural history of FTD in human cohorts. In Alzheimer’s and Parkinson’s, studies such as DIAN, AIBL, ADNI, and PPMI are showing how biomarkers change over time and how to quantify disease progression. This has helped galvanize industry, and they want the same for FTD. “We have to have longitudinal human data. That is our key ‘ask’ to be able to run successful trials,” Philipp von Rosenstiel at Biogen in Cambridge, Massachusetts, told the FTSG. Biogen is developing a preclinical anti-tau antibody.
This sequence of events is how scientists theorize FTD might develop. GENFI and LEFFTDS are generating empirical data to test this staging model. [Courtesy of Jonathan Rohrer, UCL.]
The most advanced project is the Genetic FTD Initiative. Inspired by DIAN for autosomal-dominant Alzheimer’s, GENFI started in 2012 as a multicenter consortium to gather carriers of pathogenic mutations in the progranulin, tau, and C9ORF72 genes and their relatives into a cohort. The goal was to track their disease process with standardized assessments of biomarkers and clinical symptoms, including standardized cognitive tests across European languages. “With GENFI1, a key point was to see if we would be able to do this,” Jonathan Rohrer of University College London told the audience. They were, and the result was a standing project among 13 centers in Europe and Canada that is beginning to order biomarkers across preclinical and clinical stages of disease. Cross-sectional data from a data freeze are published (see Nov 2014 conference news; Rohrer et al., 2015). When GENFI-1 ended in spring 2015 and GENFI-2 started, the scientists had collected baseline data on 365 participants and 148 follow-up visits, Rohrer said.
April 2016 marks year two of GENFI-2, which has expanded to 27 centers in Europe and Canada and is currently enrolling for a target of 600 participants coming for three visits. Some GENFI-1 participants are continuing into this next phase of the project. Thus far, 303 are enrolled, with the largest fraction being progranulin families. Because GENFI-2 can build on the infrastructure and protocol of GENF1, the researchers can now focus more on trial preparation. Besides creating a trial-ready cohort, their goals include trying to pin down biomarkers that indicate the best time to interfere therapeutically, as well as progression markers to measure whether a drug might be working, Rohrer said. GENFI will make its data broadly available for the field to study, Rohrer promised.
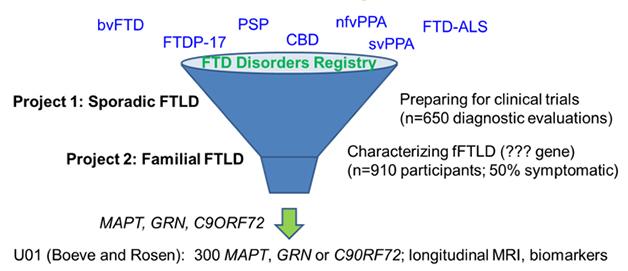
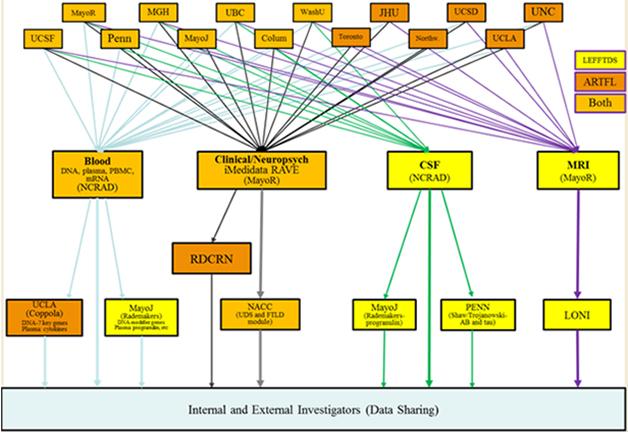
Separately, in cities across the United States plus Vancouver and Toronto, two interconnected initiatives started up in spring 2015 to accomplish much the same goals. Called ARFTL and LEFFTDS, they are described in detail in this Dec 2014 conference story.
In its twin efforts to prepare participants for upcoming therapeutic trials and discover new familial cases, the ARFTL initiative welcomes people with all forms of FTD, both sporadic and hereditary. [Courtesy of Adam Boxer, UCSF.]
This image captures how participating institutions coordinate the components of the ARTFL and LEFFTDS FTD research initiatives. Different centers house repositories for storage of blood and CSF samples, MRI scans, and clinical data. [Courtesy of Brad Boeve, Mayo Clinic, Rochester.]
ARFTL’s leader, Adam Boxer of the University of California, San Francisco, also chairs the Frontotemporal Dementia Treatment Study Groups' steering committee. In D.C., he told the audience that ARTFL is broader than GENFI and LEFFTDS in that it aims to find out how best to run therapy trials not just in genetic but also in sporadic forms of FTD. These still constitute the majority of cases, and in most of those clinicians do not have the advantage of knowing which protein pathology drives a given person’s disease.
Boxer noted that one big priority was to increase the number of participants for trials across FTD with the help of a registry. After initially exploring the existing Rare Clinical Diseases Research Network (RCDRN) for this purpose, the investigators opted instead to custom-build an online tool called the FTD Disorders Registry. Led by Dianna Wheaton, a genetic counselor who previously directed the Southwest Eye Registry in Dallas, the FTD Disorders Registry will be a resource for ARFTL/LEFFTDS, GENFI, and biopharma companies conducting clinical trials. Importantly, it will remain independent of any given research network, trial, or company. Funded by FTD charities such as AFTD, the Bluefield Project, and the Tau Consortium, this registry will be a tax-exempt LLC designed in such a way that its participants own their data, Wheaton told the group.
People with any type of FTD, as well as their family members and caregivers, will be invited to join and enter data. This will be a contact registry that will do its own research via surveys and focus groups. It partnered with the Alzheimer’s Prevention Registry and its vendors to take advantage of the APR’s software design and marketing approaches, which have brought APR membership to 214,000 far. The partnership with the APR will allow the FTD Disorders Registry to learn from GeneMatch, a genotyping program the APR is piloting to help match people to therapeutic trials that require genetic information for recruitment. The FTD Disorders Registry will dip its toes into launch on May 13 at the AFTD Annual Conference in Minneapolis, offering access into the contact registry at that point; it will launch with full functions such as registration for research participation this June, Wheaton said.
ARTFL/LEFFTDS will also use resources already available through existing NINDS programs. One is the ongoing Parkinson’s Disease Biomarker Program. The PDBP already contains data and access to biosamples on 44 participants with PSP (see PDBP data), Sutherland told the audience. In addition, the PDBP features a data-management tool that the FTD Disorders Registry and ARTFL/LEFFTDS will use to create GUIDs, aka globally unique identifiers, for their participants. Like a social security number, a GUID enables tracking of people across studies or sites, such that an individual person’s clinical, genetic and other biological data can be linked. Biosamples collected under ARTFL/LEFFTDS are stored within NCRAD, the National Cell Repository for Alzheimer’s Disease. At present, a total of about 7,000 aliquots of CSF, blood, and RNA are at the cores and available for study, Sutherland said. More broadly, NINDS is building out other existing research resources to better accommodate FTD.
In its first year, ARTFL has enrolled 260 participants across the FTD spectrum, Boxer told the audience. For its part, LEFFTDS thus far has enrolled 39 people with a pathogenic tau mutation, 20 of them presymptomatic carriers, Brad Boeve of the Mayo Clinic, Rochester, told the audience. From across 13 pathogenic progranulin mutations, 55 people have signed up, 34 of them presymptomatic, and the group with C9ORF72 repeat expansion currently numbers 48 participants, 30 of them presymptomatic. Many more kindreds are known to scientists, though not all of them are published in the literature, Boeve noted. Some kindreds are quite large; for example, one carrying the N279K tau mutation numbers more than 320 members, of whom eight have joined the study, he said. Showing examples of large pedigrees, Boeve said that many families are unaware of the extent of the illness in their kindred, and that many individuals are not yet identified.
Carriers of pathogenic mutations located across the length of the progranulin gene have joined the LEFFTDs study of genetic FTD. [Courtesy of Brad Boeve, Mayo Clinic.]
The families who were previously known to the participating sites have become keenly interested in research since they learned about AFTFL/LEFFTDS, Boeve said. Additional families have come forward, too, and Boeve expects more people to make contact once the FTD Registry opens. He noted that the bottleneck in growing LEFFTDS are the centers, not the participants. “The families are coming. There are more interested individuals than there are staff to evaluate them all, but we aim to enroll and follow all who are interested,” Boeve said.
Estimating when a given LEFFTDS participant is likely to become symptomatic is difficult at present, Boeve acknowledged. While the age at which this happens is relatively tight within families of the same tau mutation, it can range from the 30s to the 70s within progranulin or C9 mutation families. For clinical trials to measure a cognitive or clinical outcome in these people, longitudinal studies need to identify biomarker changes that flag symptom onset and estimate how fast a patient is likely to progress. Mark Forman of Merck in Pennsylvania summed up the FTD Study Group’s consensus when he said, “The heterogeneity is the main challenge in how we can run trials, and biomarkers will be critical for their success.” For news on biomarkers in FTD, see Part 2 of this series.
To help with FTD trials, Albert Lo of Eli Lilly and Company in Rhode Island proposed building a shared infrastructure to support FTD therapy development. This platform, too, could draw inspiration from Alzheimer’s, where several initiatives are underway to streamline drug evaluation. One is Eisai’s Phase 2 trial of the BAN2401 anti-Aβ protofibril antibody. This trial is innovative because it tries to cut down both the length and needed number of patients with a Bayesian design that adapts randomization into dose groups based on frequent interim analyses. This trial design, the Phase 1 data on this antibody, and the composite outcome measure developed for it, were all recently published (Satlin et al., 2016; Logovinsky et al., 2016; Wang et al., 2016). Adaptive trial elements are a prominent feature of a large European initiative to develop a standing platform for Phase 1/2 Alzheimer’s prevention trials into which multiple pharma companies are to enter their investigational drugs (Ritchie et al., 2016). And of course, the DIAN-TU pioneered the idea of a trials platform built on an observational cohort by forming a pharma consortium in 2011. DIAN-TU’s ongoing Phase 2/3 trial comparing both solanezumab and gantenerumab to a shared placebo successfully completed enrollment last December (Dec 2011 conference news; Apr 2015 conference news; DIAN-TU press release).
How about it for FTD? At least in initial public comments at this early stage FTSG meeting, Lo’s proposal drew a tepid response. Would pharma companies withhold their best investigational drugs from such a platform? That has not been the case in DIAN-TU. Would management demur? The reticence in the room prompted a plea from Howard Feldman, University of California, San Diego. “Let’s remember there’s considerable urgency to this problem, and our progress is very slow. Unless we come together around something innovative, like this adaptive trial platform, we run the risk of continued very slow progress. It may not be for every company. Maybe it is for smaller, hungrier biotechs,” Feldman said.—Gabrielle Strobel
Rohrer JD, Nicholas JM, Cash DM, van Swieten J, Dopper E, Jiskoot L, van Minkelen R, Rombouts SA, Cardoso MJ, Clegg S, Espak M, Mead S, Thomas DL, De Vita E, Masellis M, Black SE, Freedman M, Keren R, MacIntosh BJ, Rogaeva E, Tang-Wai D, Tartaglia MC, Laforce R Jr, Tagliavini F, Tiraboschi P, Redaelli V, Prioni S, Grisoli M, Borroni B, Padovani A, Galimberti D, Scarpini E, Arighi A, Fumagalli G, Rowe JB, Coyle-Gilchrist I, Graff C, Fallström M, Jelic V, Ståhlbom AK, Andersson C, Thonberg H, Lilius L, Frisoni GB, Pievani M, Bocchetta M, Benussi L, Ghidoni R, Finger E, Sorbi S, Nacmias B, Lombardi G, Polito C, Warren JD, Ourselin S, Fox NC, Rossor MN.
Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis.
Lancet Neurol. 2015 Mar;14(3):253-62. Epub 2015 Feb 4
PubMed.
WANTED: Biomarkers for Drug Trials in Frontotemporal Dementia
At a recent workshop hosted for the Frontotemporal Dementia Study Group on March 31 and April 1 in Washington, D.C., researchers in academia, industry, and regulatory agencies universally agreed on one thing. They all want biomarkers to conduct clinical trials. Ideally, a whole bunch of them. Humbled by the hunt for Alzheimer’s drugs, scientists want FTD markers for several reasons.
For one, they need help picking the right patients for any particular trial. “It is critical to ascertain that patients in your trial have the disease you think they have. It was a big surprise to discover in the bapineuzumab trials that 30 percent of the patients did not have the disease the drug was designed to treat. Hopefully this will not be a problem in FTD,” said Geoffrey Kerchner of Genentech, South San Francisco. For another, they want pharmacokinetic and -dynamic markers to learn exactly what their drug does after they give it to people. “We must know if the compound was on board and hit its target. That is key. We must have that data so we can drop a target,” said Christoph Wiessner of Asceneuron. This biotech company in Lausanne, Switzerland, is hoping to start testing a small-molecule drug targeting a post-translational modification of tau in 2017.
From the Test Tube: Fluid Markers in FTD
Several candidate biomarkers exist, but how reliable are they? Kaj Blennow of the University of Gothenburg, Sweden, updated the group on the status of fluid markers in FTD. Cerebrospinal fluid markers for the molecular pathology of Alzheimer’s disease, i.e., Aβ42 and tau, have come a long way and are even approaching certification as clinical-grade diagnostic aids. Alas, researchers have known for at least a decade that neither total tau nor phospho-tau assays work in FTD (Hampel et al., 2004; Olsson et al., 2005). This was a head-scratcher—after all, tau pathology is the hallmark of many FTD disorders. Current tau assays are based on antibodies that recognize the protein’s mid-domain, and have been widely thought to measure “total” tau levels. However, recent mass spectrometry studies have shown that human CSF contains little full-length tau protein, but rather a range of N-terminal and mid-domain tau fragments. This raises the possibility that there are FTD-specific tau species in human CSF. If so, further mass spectrometry studies may identify fragments that could serve as the basis of FTD-specific tau assays, Blennow said (Portelius et al., 2008; Meredith et al., 2013).
The new darling in the AD biomarkers field—neurogranin—also appears to be a non-starter in FTD. Neurogranin has long been known to be a component of dendritic spines, and new antibodies to it have led to tests that are rapidly gathering evidence supporting neurogranin as a CSF marker for synaptic degeneration in Alzheimer’s. Alas, the few studies that have thus far compared neurogranin across a range of dementing disorders suggest its signature increase may be specific to AD. This means the current AD fluid markers are useful indirectly, because they help exclude an FTD diagnosis, Blennow said.
Does anything work directly in FTD, then? Thus far, researchers are having more success with neurofilament light protein. NFL is a component of the cytoskeleton in large-caliber axons that course through white matter. Its concentration tends to rise in the CSF in instances of active neurodegeneration, including traumatic brain injury. NFL has been known since the year 2000 to go up in the CSF of people with FTD, but not AD, and it was rediscovered in 2014 by Adam Boxer’s group at University of California, San Francisco, and others. A comparison of 3,355 patients showed that CSF NFL increases about threefold in FTD, markedly higher than in other dementing illnesses, including AD, dementia with Lewy bodies, and vascular dementia (Sjogren et al., 2000; Scherling et al., 2014; Skillback, 2014).
The concentration of NFL in plasma amounts to less than a sugar cube dropped into an Olympic-size swimming pool. Could a blood test measure it accurately to spot FTD? [Courtesy of Kaj Blennow, U. Gothenburg.]
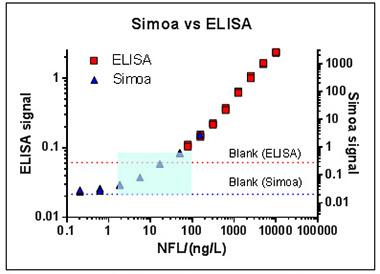
Could a blood test detect NFL? It’s a tall order. Compared to NFL’s concentration in the CSF (500 pg/ml), its concentration in plasma is but 10 pg/ml. This amounts to 3 percent of a sugar cube’s worth of NFL dissolved in an Olympic-size swimming pool that also contains 40 tons of other proteins, Blennow said. Abundant proteins such as CSF albumin would weigh in at 350 kg in the same pool.
But according to Blennow, a single molecule array (aka Simoa) assay can do it. The assay uses capture and biotin-tagged detection antibodies, as do ELISAs, but its bead-based technology and digital readout give it greater sensitivity. It measures NFL in serum down to a limit of 0.6 pg/ml, as compared to 78 pg/ml for ELISA and 16 pg/ml for Mesoscale. “You need these kinds of ultrasensitive technique to measure NFL in blood,” Blennow said.
A single molecule array assay is more sensitive than ELISA, enabling scientists to detect neurofilament light chain in serum in the vanishingly low concentrations at which it occurs in human blood (blue box). [Courtesy of Kaj Blennow.]
Further data showed that NFL in blood correlates tightly with NFL in CSF; in other words, that serum NFL reflects brain pathology. With this reassurance, Blennow and collaborators at University College London tried the new test on blood samples of various FTD subgroups. Initial analyses of GENFI and other samples are beginning to show elevated serum NFL in all FTD subgroups, particularly in FTD-ALS and in progranulin mutation carriers, Blennow said. People with more severe symptoms tend to have higher serum NFL, and their brains shrink faster. This link between high serum NFL and fast decline is beginning to emerge in GENFI, and it has been published in the much larger ADNI cohort (Zetterberg et al., 2016). For clinical trials, NFL might therefore help researchers identify patients who are likely to progress fast. NFL in blood is also elevated in some parkinsonian disorders such as corticobasal syndrome (CBS), progressive supranuclear palsy (PSP), and multiple-system atrophy (MSA). Curiously, NFL shows no marked elevation in Parkinson’s, a slower-moving disease.
“Plasma NFL is a sensitive but unspecific progression marker for neurodegeneration,” Blennow said. As a candidate progression marker, could it flag a treatment effect in trials? There is no FTD data yet, but in multiple sclerosis, the answer is yes, at least according to a study in which blood NFL levels returned to normal in patients who responded to natalizumab antibody treatment (Gunnarsson et al., 2011). If blood NFL holds up in further testing, it may obviate the need for spinal taps in FTD, Blennow said.
What about blood proteomics and metabolomics? Most studies on multiple-ligand assays have started with up to 1,200 molecules and ended with panels of 18 or less that reportedly discriminate between groups. Typically, the changes in individual ligands are minute but collectively, their change can be significant in a statistical model. The small number of patients in these studies raises a concern of data over fitting, Blennow cautioned. Biomarker studies need to move beyond validating a panel by dividing the original cohort into a training and a validation set and adjusting the panel to optimize its discriminatory power. The way toward a robust assay is to take the panel to a totally different sample of people and try to validate the original finding. “Fix your panel and test it in a new cohort. If you can do that three, four times, then you have something,” Blennow said.
What about cytokines? Blennow noted that despite the growing interest in neuroinflammation in neurodegenerative disease research, general inflammation markers are difficult to measure. Cytokines are present at low levels of 1 to 2 pg/ml and tend to hover at the bottom of their assays’ calibration curves. Blennow recommended investigators run their samples alongside samples from multiple sclerosis or a chronic infectious disease such as Lyme to gain a sense of how cytokine changes in FTD compared to those in known neuroinflammatory conditions. “You typically see a slight change in AD or FTD compared to an enormous difference in MS,” Blennow said.
From the Scanner: MRI and PET Bill Seeley of the University of California, San Francisco, compared structural and functional connectivity MRI. The former measures atrophy in particular brain areas. Its high signal-to-noise ratio and resolution make it attractive in FTD for making clinically defined subgroups more homogeneous, or for monitoring disease progression. Alas, the researchers also noted limitations of structural MRI as a potential outcome measure, including potential confounding fluid shifts in the brain. Fluid shifts have repeatedly produced puzzling results in some Alzheimer’s trials, where the brain temporarily shrank more in treated patients than in those on placebo (e.g., Jul 2004 conference news). Moreover, while researchers believe structural MRI might detect large effects of disease-modifying drugs, they do not expect an atrophy signal with symptomatic drugs.
All this is why high hopes have rested on imaging methods that measure brain function. Most recently, FTD researchers have focused on resting-state fMRI. In particular, a method called seed-based fMRI can illuminate the specific network of functionally connected brain areas that degenerate in a given FTD disorder, all of which are network diseases, said Seeley. For example, by using the dorsal midbrain tegmentum as a seed region of interest in healthy older people, scientists can see coordinated firing of a network that is a striking match to the atrophy pattern in PSP, and whose connectivity becomes degraded as PSP progresses, Seeley told the audience.
Alas, while elegant and promising, resting-state connectivity MRI is not ready for prime time, the scientists agreed. It is “noisier” than structural MRI and quite sensitive to head movement, a frequent problem when scanning people with a behavior disorder. Connectivity MRI is also sensitive to factors that subtly influence a person’s mental state, even simple things such as whether the patient had coffee prior to the scan.
Then again, older methods of imaging brain function, such as FDG PET, do pick up relatively rapid increases in brain function when a symptomatic drug works. Task-based fMRI tests are being evaluated in some trials. While none are robust and validated yet, new drugs such as AGB101 are targeting network excitability and will require a functional MRI readout. In D.C., investigators in both academia and industry emphasized that they need dynamic biomarkers that can tell them sooner than currently used readouts whether the drug at hand warrants a larger study or should be scrapped. Seeley noted that his research monitoring symptomatic FTD patients indicates structural MRI can flag decline over eight weeks. “We should develop these measures further,” agreed Brad Dickerson of Massachusetts General Hospital, Boston.
Summarizing PET imaging in FTD, Dickerson first noted that amyloid PET is already useful in dementia clinics to rule out Alzheimer’s disease. As an example, he told the case of a 62-year-old academic physician who one day tried to give grand rounds and sat down after 10 minutes, thinking he was finished. Initially he was thought to have AD, but his PiB scan came back negative, prompting more tests. His frontal lobe turned out to be hypometabolic on FDG PET and shrunken on structural MRI. He carried a progranulin mutation.
Tau PET is what everyone is is waiting for impatiently. With tau PET, researchers hope to see not only that patients in a trial of a tau-targeted drug have neurofibrillary tangles, but also whether the drug halts their spread. “For example, if you can show that you block progression of tau in PSP, we would be extremely excited as a company,” said Mark Forman of Merck in Philadelphia.
Alas, it’s early days. The best-studied tracer thus far, T807/AV1451, works well in Alzheimer’s but not FTD. “T807 is not the tracer we will use in FTD in the future, but it can teach us a lot along the way,” Dickerson said, to nods around the room. As Dickerson’s and other groups are scanning FTD patients they suspect of having tau pathology—for example symptomatic and presymptomatic tau mutation carriers, people with PSP—they are indeed seeing a T807 signal in expected areas of atrophy. The trouble is that they also see a signal in people whose FTD is likely due to TDP-43. More trouble: The T807 signal seen in living patients does not fully match results of autoradiography and other methods of postmortem tissue validation.
This may seem like a setback after the field had greeted tau PET with enthusiasm, but Dickerson said more research will surely clarify the binding properties of T807. What’s more, a handful of other tracers are nipping at its heels and will make for a good comparison. For one, the THK5351 tracer coming out of Tohoku University in Japan, looks promising, Dickerson said. Roche, Piramal, and Genentech all have started evaluating their tau tracers in people. Even Merck, a therapeutics giant that has no diagnostics business, has decided to make a tau PET tracer.
In D.C., Forman told the FTD Study Group that his company is developing the Merck tau PET tracer together with researchers at MGH in a joint project funded by the Alzheimer’s Drug Discovery Foundation. This tracer also has entered human trials, and first results are expected later this year. “It is new for us to develop an asset publicly and collaboratively. We do it because we want a high-quality tracer that will be available to the community and supports GENFI, ARTFL, and LEFFTDS. To us, these initiatives are moving the needle,” Forman said.
Besides tau, PET tracers are coming up for other targets, for example histone deacetylase (Strebl et al., 2015). While the neuroinflammation tracer PBR-28 is showing signals in neurodegeneration (see Zurcher et al., 2015), this ligand requires genotyping patients to understand their PET signal relative to control. The FTD Study Group agreed that the field needs to develop new imaging and fluid markers of neuroinflammation, ideally specific markers for well-characterized targets.
Over and over in discussion, industry, academic, and regulatory researchers called for rigor in biomarker development and for learning the bitter lessons of AD. They emphasized that any drug moving forward should have strong evidence of target engagement. “For this, we have to have quick response measures,” said Philipp von Rosenstiel of Biogen in Cambridge, Massachusetts.
Currently in Alzheimer’s disease, companies are running large, long Phase 3 trials to look for small signals. This is in part because there are no validated outcome biomarkers and in part because companies are reluctant to give up candidate drugs that already cost so much time and money to move through Phase 2. The FTD Study Group hopes that with better biomarkers, FTD trials will more efficiently tell researchers whether the investigational drug at hand works. This is especially critical in a set of rare spectrum disorders, where trials will have to stay small. “We have many examples in AD where we said we can power up and do a muscular trial and get over these problems. But that is wrong. We need to have biomarkers so we can interpret trial results,” said Howard Feldman of the University of California, San Diego, who took over the Alzheimer’s Disease Cooperative Study.
Most people with FTD are younger than late-onset AD patients, hence they have fewer age-related morbidities that muddy the disease picture and make them prone to side effects. But just like in AD, intervening early is important and, at least in sporadic cases, that is impossible without biomarkers. “We are all in line with the notion that by the time a patient comes to clinic and a simple interview makes clear they have FTD, it may be too late. Hopefully we can learn to diagnose patients when their clinical symptoms are very subtle. The field is clearly aligned around that goal,” Kerchner said.
All in all, the upshot of the day was that some FTD biomarkers are already useful in exploratory trials and should be embedded in all FTD trials to generate data, but as of 2016, none are close to receiving formal regulatory qualification for a particular context of use. To learn about the regulatory discussion, see Part 3 of this series.—Gabrielle Strobel
Regulators Tell Frontotemporal Dementia Community: We Play on Your Team
On April 1 in Washington, D.C., a meeting convened by the FTD Treatment Study Group offered an assembly of academic and industry scientists, funders, representatives of patient organizations, and caregivers the rare opportunity to collectively pick the brains of senior scientists from the Food and Drug Administration and the European Medicines Agency. How should we develop therapies for the rare and heterogeneous set of diseases that make up frontotemporal dementia, they wanted to know?
As no drugs are approved to treat FTD, none have undergone a formal regulatory process. No framework exists to guide the various drug development programs that are sprouting up across the United States and Europe. The EMA in January 2016 invited public comment on a draft guideline for drug development in Alzheimer’s disease and other dementias. However, it does not clarify concrete questions such as what role fluid or imaging markers can play as supportive or primary evidence for the registration of drugs developed in small, heterogeneous populations of patients. The FDA in 2015 issued a draft guidance on rare diseases, but it does not address FTD specifically at all. This absence of a status quo is both a challenge and an opportunity, said Philipp von Rosenstiel of Biogen, Cambridge, Massachusetts. The conversation in D.C. helped scientists gauge—and try to influence—how regulators currently think about these devastating forms of dementia.
In previous discussions, scientists had laid out how FTD differed from Alzheimer’s, for example by its bewildering lack of correspondence between an underlying pathophysiology and its phenotypic expression (see Part 1 of this series). They established that FTD is distinct from AD in its biomarker profile, and that much more work is needed there (Part 2). In this discussion, the scientists peppered regulators with questions to get a feel for how their own preclinical or clinical programs might be perceived. Many drug sponsors meet with regulators privately for advice based on data on a given drug in development, but in group conversations such as this, mentioning specific investigational drugs is largely taboo. Instead, the dynamic tends to be one where regulators answer questions with general principles, generating nuanced “it depends” answers that don’t pin down the regulator but nonetheless offer valuable insight into how regulatory thinking evolves along with advances in the field.
Maria Isaac of the EMA, with Billy Dunn and Nick Kozauer of the FDA, urged FTD researchers to absorb the lessons of failed trials in AD. Above all, develop biomarkers, they said. “You will find us very receptive to their use,” Kozauer said. Apply biomarkers to characterize the natural history of FTDs. Use them to ascertain that each participant has the underlying etiology the drug is targeting. Regulators recommended that scientists develop creative outcome measures, such as individualized ones, but urged the field to stay laser-focused on showing that a drug effect is meaningful to the patient.
If this sounds like a high bar, the regulators also emphasized that they can be flexible and that they want to see therapies succeed as much as everyone in the room. “Some of us have treated these patients as clinicians. We are very aware these are horrible diseases,” Kozauer said. “Together, we have a fantastic opportunity here. We have talked internally about the important need in FTD and are enthusiastic about greater attention to this area. By learning from AD, we should be able to bring greater efficiency to the process,” Dunn said. Most of all, regulators emphasized the need for individual players to avoid working in silos. “Share resources and data. It gets us there faster,” Dunn said.
Sometimes academic or industry scientists blame regulators for slowing innovation in drug development, but Dunn disputed this notion. “There is no disconnect between academics, industry, and regulators. We are all on the same team. We each play different roles, but we are here today to push for robust progress toward this goal,” Dunn said. Likewise, Isaac told the group,” I am happy to discuss your programs with you.”
Besides Biogen, biopharma companies that dispatched researchers to this meeting include A&G Pharmaceutical, Alector, Asceneuron, Bristol-Myers Squibb, Cogentis, Cydan, Forum, Genentech, Janssen, Lilly, Merck, Novartis, Qanterix, TauRx, as well as several contract research organizations. For paraphrased excerpts of the main discussion points, see Q&A below.
Q: What is the single thing that would propel drug evaluation in FTD the fastest?
A: Finding biomarkers that allow you to positively demonstrate the presence of the underlying hallmark lesions in vivo, and to discriminate between the etiological types of FTD. You need tau biomarkers, TDP-43 biomarkers, FUS biomarkers.
Q: How do you evaluate drug risk in a fatal neurodegenerative disease where there is no therapy?
A: We place those conditions into a special category. We understand the tolerance for risk is very high. That said, we need to be sure that we can contextualize and describe for the patients in a responsible way what the risks and benefits are. It’s tough to give a bright line because we consider this on a case-by-case basis. We need to see the evidence but assure you that we think hard about this.
Q: Do you think differently about risk tolerance in Phase 1 than registration?
A: Yes. In an approval decision, where we have efficacy data and can consider risk versus benefit, we can be flexible on how many people we need to see evidence on. According to the International Council of Harmonisation, it can be as few as 100 patients exposed for a year. Earlier in development, we are primarily concerned about risk because you know very little about the drug’s benefit. We can be somewhat flexible in how many exposures, and how long, you need in order to go forward. But we have had cases where a drug seemed safe early on and with more exposures bad things did happen.
Q: Long toxicology studies really slow things down. Meanwhile people are suffering.
A: We understand you want to find a streamlined approach. We have specific experience where, in an effort to accelerate, we allowed shorter-than-normal toxicology studies to support long-term human studies. That can work, but we have also had situations where toxicity emerged in longer-term animal studies after the drug was already in humans, and was so severe that the clinical program had to be stopped. We are eager to bring drugs forward but are aware of both sides of the issue.
Q: Does previous human exposure count?
A: Yes. It is extremely important. If adequate, that data can be used to support clinical studies in the absence of certain pre-clinical studies.
Q: How do you balance an educated, informed population against a paternalistic approach? How important is the patient’s knowledge and decision?
A: We favor an informative approach. We have heard from this patient community that they want information and want to able to make a decision. We do not say this is right and wrong for you.
Q: To illustrate the desperation we are facing: Those of us who have done trials in FTD hear both patients and families say it would be better if the person died more quickly rather than slowly. They cannot deal with what the disease does to them anymore. They say if a drug has serious side effects for the patient, that could not possibly be worse than what they are living already.
A: We understand and have great hope for the trials being started in these diseases.
Q: What endpoints could support approval? Is it necessary to demonstrate an effect on a functional or global outcome measure, or could a composite scale suffice?
A: It could. The bottom line is you have to do something meaningful for the patient—meaningful both in terms of the content, i.e., what the scale assesses, and in terms of how it assesses it. In FTD we know so little that we will not be prescriptive about recommended endpoints. Remember that even when we have considerable experience with an endpoint, the FDA or EMA remain open to proposals for other endpoints.
Q: Please elaborate.
A: Our openness about endpoints is true for many diseases, even the ones where we know much more. In those diseases we may ask you to include the assessment that we know to be the innovative one, but here we do not even have that. As you choose endpoints, our advice is to keep it simple. We really just want to see something that is meaningful for the patient. That is what we are after. We are very open to how you do that.
Q: What makes a scale meaningful?
A: Some confusion may stem from the limited experience with using composite scales in predementia trials as a single endpoint. We discuss this in our draft guidance for early Alzheimer’s. For example, the CDR is a composite in a sense. Its appeal is that as a clinician-rated scale it assesses cognitive domains through the lens of how a person functions. It is not just neurometrics out of context. If you measure neuropsychology without life context, then you are in trouble. In a trial you need to assess how someone is functioning. Do not cobble together a composite with some pure neuropsychology elements and some questions about daily activities. Build a composite where each individual element by itself is clinically meaningful.
Q: Is the Progressive Supranuclear Palsy Rating Scale (PSP-RS) sufficient as a global outcome measure for PSP?
A: A general caveat here is that some of these scales may be valid for following clinical progression but may not be ideal for use an as endpoint in clinical trials. We see promise in the PSP-RS but have very little experience with it. It may need to be adapted somewhat to serve as a trial outcome measure. It could be pared down to its more meaningful parts. We are happy to discuss the specifics of measures that sponsors propose.
Q: Do we need a co-primary? For example, would language be sufficient in aphasia?
A: If you assess an important cognitive domain like language in a way that shows the drug improves the person’s ability to function, then that is sufficient. But your language measure has to be meaningful.
Q: How about wearable devices to record outcomes?
A: We are very open to that. You will find us more flexible than you think. The clinical meaningfulness of what the device measures is where you will get scrutiny from us.
Q: We have been working on patient-reported outcomes for corticobasal degeneration. Many CBD patients have poor insight. They cannot communicate outcomes or improvement, so it is the caregivers who can tell.
A: We encounter that all the time. We understand that in the early stage, some people retain insight but the variability of this can cause problems in a trial. For example, assume the drug worked. Patients who take the drug may retain insight into their deficits while patients on placebo may lose insight and incorrectly perceive that they are doing fine. In that situation it might appear that patients taking the drug are doing worse than the patient taking placebo.
Q: And this means?
A: That despite our fervent belief in the patient’s voice, cognitive impairment can affect the reliability of that voice. We are engaged with the PRO consortium on this topic.
Q: How do you view volumetric MRI as an outcome marker versus cognition or a clinical measure?
A: As potential supportive data.
Q: Could accelerated approval based on a biomarker related to the disease mechanism be applied under subpart H? For example, one genetic cause of FTLD is haploinsufficiency of the progranulin gene, which causes low progranulin protein levels in brain. Could approval be pursued on the basis of restoration of CNS progranulin levels combined with a reduction for a fluid biomarker of disease activity, e.g., neurofilament light chain?
A: It is very tempting to jump in on this. At this point we do not understand the pathophysiology of progranulin and NFL well enough to base approval on them. Biomarkers will be incredibly valuable here, it’s just that we need to learn more first. Consider amyloid as a cautionary tale. There was interest in using amyloid as a surrogate for approval. It seemed so logical at the time. But we needed to learn more and based on what we know now, the relationship between amyloid and clinical benefit is not clear.
Q: What sort of knowledge would make you feel more comfortable? What can we do short of 10 years of study?
A: Understand the pathophysiology of the biomarker, and link a clinical benefit to change in that marker.
Q: Our community is excited about NFL because many labs have similar findings, and NFL makes sense in terms of neurodegeneration. What more do you need to see on it?
A: We need to have data to deduce that reducing NFL is reasonably likely to predict meaningful benefit. We can’t provide a specific answer without more data, but it seems based on the breadth of our experience that additional work should be done. In general, do not think that accelerated approval implies a lower standard. The evidence of an effect on a biomarker outcome that is reasonably likely to predict a clinical benefit must be robust. If you can obtain a clinical benefit in a well-designed study, that is what you should do. It does not help to invoke accelerated approval just because. True surrogacy is very hard to achieve.
Q: Let’s say we have data to show low progranulin causes disease, and that NFL levels rise with disease. If we then see with a drug an increase in progranulin and a reduction of NFL, I’d say that’s great. Let’s see what happens. If not subpart H, can we then invoke expanded access?
A: We are willing to consider proposals for expanded access. Or in Europe you can go for conditional approval.
Q: What if the primary endpoint missed statistical significance but goes in the right direction, and NFL goes in the right direction, and FDG PET goes in the right direction?
A: You talk about consistency of effect on different biomarkers that are all in the pathway of disease. This can help a study as part of the totality of data.
Q: Do genetically defined labels extend to all clinical phenotypes of a given mutation? In other words, if we define a label based on the presence of pathogenic C9ORF72 expansion, would the label extend from C9ORF72 ALS to C9ORF72 ALS-FTD and C9ORF72 FTD?
A: We try to generalize the results as far as is scientifically reasonable, but you need to show that the mutation is pathogenic for each phenotype in question.
Q: Is the answer the same for pathology? For example, if you have a drug that treats, say, a tauopathy?
A: It depends on what you know about the conditions. Potentially yes, but it is complicated and you need to consider whether the tauopathy in the condition you want to extend the label to accounts for as much of the clinical disease as it does in the original condition and that treating the tauopathy is beneficial.
Q: With the different mutations, pathologies, and phenotypes, the number of available patients for any kind of homogeneous patient group is small. How do you deal with that?
A: There is no minimum number. We can’t get too specific as the information and your outreach are evolving, but we understand you can only do what you can do. Just know that a limited pool of patients is not prohibitive for drug development. We have approved drugs for small populations.
Q: Can a PSP clinical trial performed in an adaptive platform study serve as a registration trial intended to provide primary efficacy evidence to support approval?
A: With the important caveat that the devil is in the details, the answer is yes. Primary efficacy data can be generated from such an approach.
Q: How about the pooled placebo used in such trial platforms?
A: There are details to consider, and we all know placebo groups can go bad in lots of ways. But yes, it can be acceptable. There are efforts underway that use pooled placebo. You do want high throughput and to streamline trials. We encourage that. If you are concerned that you can only get exploratory evidence with that, that is not the case.
Q: In PSP, can a single clinical trial be sufficient for approval?
A: Since the question is “can,” the short answer is “yes.” Typically, the evidence comes from two studies. But if a single trial is large, shows a big effect, survives multiple sensitivity analyses, hits multiple domains etc., then it can be sufficient. We have guidance on that.
Q: Can we prospectively use domains that are abnormal at baseline in individual subjects, as opposed to a broad spectrum scale for everyone?
A: Yes. We are open to patient-specific outcomes. If you individualize endpoints and specify that in the trial, then that could be a useful approach. It is intrinsically attractive as you want to treat the thing that is bothering that patient the most. We hear back from sponsors that patient-specific outcomes can be tricky to operationalize. You’ll have to work out a lot of details about the statistical analysis plan and other aspects. Ask us and we will advise.
Q: With this approach, do you still also want a unifying global outcome that is applicable across all these subtypes? The spectrum of symptoms is so wide.
A: That depends on the case. Not necessarily. One potential way to deal with the heterogeneity is to develop a disease-independent measure that captures the totality of how a patient is doing.
Q: What preclinical data will the FDA want to see for FTD caused by progranulin mutations? There are no good disease models yet.
A: We often get presentations of drug efficacy in animal models. Be aware that that may not be a large regulatory consideration in drug development. From our standpoint, animal data are most often used to inform safety considerations. Animal efficacy is often important to sponsors to make decisions about moving forward.
Q: In an ideal world, what evidence would you want?
A: You would show us this: “We caught these people bound to develop this disease early. Here is how they are already changing preclinically, here is what is going to happen to them in roughly this amount of time, here is how our therapy has affected these disease-related biomarkers, and here is why that is reasonably likely to delay people’s progression.” Bring us these data. We are in the same boat, and don’t want to be too late for these patients.




Comments
No Available Comments
Make a Comment
To make a comment you must login or register.