Past Webinar
The Cell Cycle and Alzheimer’s Disease—Let's Unite for Division!
Quick Links
Introduction
Inez Vincent led this live discussion on 7 August 2002. Readers are invited to submit additional comments by using our Comments form at the bottom of the page.
Transcript:
Live discussion held 20 May 2002 at 12 noon.
Participants: Inez Vincent, Zsuzsanna Nagy, Rachael Neve, Karl Herrup, Nathaniel Milton, Mark Smith, Ben Wolozin, Yuzhi Chen, Kung Ping Lu, June Kinoshita, Gabrielle Strobel, Jan Hallows, Robert Bowser, Douglas Feinstein, Craig Atwood, Ken Maiese.
Note: The transcript has been edited for clarity and accuracy.
Gabrielle Strobel: Hi everyone, and welcome to today’s chat on a fascinating hypothesis in Alzheimer's disease. I am managing editor at the Alzheimer Research Forum and will be moderating today. Why don’t we start by asking Inez Vincent to restate her hypothesis and say what is most needed now to further test it.
Inez Vincent: I would like to start by welcoming everyone to this discussion on the cell cycle and Alzheimer's disease. Let’s cycle!
The basic hypothesis is that entry of post-mitotic neurons into the cell cycle, followed by an uncoordinated progression through the cycle, leads to degeneration of neurons in conditions such as Alzheimer's disease. As far as advancing the theory, well, this is the million-dollar question! There are several ways to attempt it:
1. Better studies of human brain, both normal and those afflicted by other degenerative disorders, using rapid post-mortem tissue.
2. Development of models based on findings in human brain.
Zsuzsanna Nagy: One of the problems with rapid post-mortem analysis is that, most of the time, the brain is not normal. The same goes for biopsy tissues.
Karl Herrup: How does the mitotic re-entry fit in with other theories? For example, what about the inflammation hypothesis?
Nathaniel Milton: Or what about the amyloid hypothesis? How does Aβ fit in?
Mark Smith: Without tying the cell cycle in with our current understanding, we don’t have a clue!
Zsuzsanna Nagy: I think the mitotic re-entry is not restricted to Alzheimer's disease. I think it might be a common path to degeneration that occurs in several different diseases.
Karl Herrup: I agree with Zsuzsanna.
Mark Smith: Zsuzsanna, then do you think cell-cycle re-entry is a late event in neurodegeneration, or at least a non-specific event?
Zsuzsanna Nagy: I don’t think it is late, I just think that mitotic re-activation on its own is the way neurons degenerate in general. What follows after re-entry turns the various conditions into specific diseases.
Ben Wolozin: OK, so we have three possible suspects: inflammation, Aβ, and presumably oxidation. What is the temporal progression? Mark, don't you think the cell cycle events come first?
Mark Smith: Ben, yes I do, and have evidence that the cell cycle comes first (at least before your proposed initiators).
Rachael Neve: Mark, what's your evidence that the cell cycle comes first?
Mark Smith: Rachael, we and others (Inez, Karl, etc.) have looked at temporal progression, and cell cycle events are early.
Karl Herrup: Inez, I like the track you're on. I've been thinking, though, that there may be post-mortem events that skew the details of the message we're after (and that means I agree with Zsuzsanna).
Inez Vincent: Our studies of different degenerative disorders have revealed that entry into the cell cycle is not a universal mechanism for degeneration.
Karl Herrup: Of course it's not universal, but it's not unique to AD, either.
Inez Vincent: There is specificity in NFT and other tau-associated disorders.
Gabrielle Strobel: Zsuzsanna, several groups, including yours, have found cell-cycle aberrations in peripheral lymphocytes in AD patients. Is this connected to AD? Or is this just a reflection of older people’s increasing risk of loss of checkpoint control?
Zsuzsanna Nagy: Interesting question, Gabrielle. We are working on it. However, since my control and AD groups were age-matched, the effects seem to be AD-related rather than a consequence of age.
Ping Lu: I think that the cell cycle might be related to tauopathy.
Inez Vincent: That's an interesting idea Ping: Perhaps tau is the most vulnerable substrate?
Zsuzsanna Nagy: I must disagree with Inez. Temporal lobe epilepsy is not a tauopathy and has no AD-like features but neurons still degenerate via the cell cycle.
Gabrielle Strobel: Ping, could you give us an example of how conformational changes after phosphorylation fit into the cell-cycle hypothesis of AD? After phosphorylation of certain Ser/Thr-proline bonds, Pin-1 changes their conformation and then what happens? Could you explain your hypothesis?
Ping Lu: I think that Pin1-induced conformational changes are important for the cell cycle, oncogenesis, and neurodegeneration, such as tauopathies.
Karl Herrup: But I'm with Mark. You can pick up cell cycle changes in the absence of phopho-tau.
Robert Bowser: I agree with Mark and Karl. Cell cycle changes can occur independently of tau phosphorylation.
Zsuzsanna Nagy: It appears that Karl and I agree on the phospho-tau and cell cycle relationship (or the lack of it).
Ping Lu: Tau phosphorylation is a result of cell cycle, through mitotic activation.
Zsuzsanna Nagy: Ping, you’ve got it right, I think.
Mark Smith: I think the cell cycle has two direct consequences: phosphorylation and oxidative stress. Both of these events lead to accumulation of phospho-tau and this (somehow) protects cells. Remember that most neurons that die have no phospho-tau!
Craig Atwood: Yes.
Karl Herrup: Do we think that phospho-tau is a necessary consequence of cell cycle activation?
Nathaniel Milton: If the cell cycle comes first, then phosphorylated tau and Aβ could be a close second. These may not be essential for all types of neurodegeneration, but maybe for AD.
Ping Lu: Pin1-induced conformational change is required for dephosphorylation of many MPM-2 epitopes, which may be linked to tau hyperphosphorylation.
Inez Vincent: I would have to refer to Thomas Arendt’s idea of neuronal activity-driven processes that may lead to cell cycle activation. He has suggested that degree of plasticity (a good thing) might eventually lead to activation of the cell cycle and death (a bad thing).
Ben Wolozin: I would like to throw out a simple hypothesis that might explain much of these phenomena. Intracellular accumulation of Aβ (or tau) leads to proteasomal inhibition. All of the cell cycle-regulated proteins are also controlled via proteasomal degradation. Hence, as proteasomal inhibition increases, the activity of the cell cycle regulators increase and the neurons go into a state reminiscent of mitosis. Thus, anything that inhibits the protease might cause this.
Inez Vincent: Any idea, Ben, why this might not happen in the transgenic mouse models that produce abundant Ab?
Ben Wolozin: These animals produce lots of Aβ, but it is extracellular, and the neurons are not damaged until late.
Jan: Ben, but what leads to the initial activation of the cell cycle prior to the accumulation of tau or Ab?
Ben Wolozin: Jan, I think the simplest explanation is that small Aβ oligomers or oxidative stress cause the proteasomal inhibition.
Zsuzsanna Nagy: Ben, how do you explain the cell cycle-related death in neurodegenerative disorders that do not have tau or amyloid accumulation?
Ben Wolozin: Many things cause proteasomal inhibition—oxidative stress, aggregated proteins. Hence, one can get to the same phenotype from many directions.
Karl Herrup: I like the proteasome idea in general, but wouldn't this kill a neuron pretty fast?
Mark Smith: I think the inhibition of the proteasome comes secondary to oxidative stress.
Zsuzsanna Nagy: I second that, Mark.
Robert Bowser: Inflammatory responses may also activate cell cycle kinases.
Zsuzsanna Nagy: can you elaborate on that, Bob?
Robert Bowser: Inflammation activates signaling pathways that can regulate the activity of kinases involved in cell cycle regulation.
Inez Vincent: Clearly, proteasome activity is critical for cell cycle progression.
Douglas Feinstein: Could there be a relationship between this proteasomal inhibition, chronic NFkB suppression, and the activation of the cell cycle?
Rachael Neve: I think you're on to something there, Douglas.
Douglas Feinstein: Perhaps this brings together aspects of inflammatory activation and the role of NFkB in promoting (or inhibiting) cell proliferation?
Ben Wolozin: Douglas, the connection with the proteasome and NFkB is easy, since IkB, which inhibits NFkB, is regulated by the proteasome. However, one could also easily explain NFkB effects independently of the proteasome because there is so much inflammation in AD.
Nathaniel Milton: Ben, isn't AD the phenotype arrived at from many directions?
Ben Wolozin: Nathaniel, I am not proposing that proteasomal inhibition causes AD, just the specific hypothesis that proteasomal inhibition could explain changes in cell cycle proteins.
Douglas Feinstein: Ben, I absolutely agree. But there could also be long-term, chronic buildups in some other NFkB-related proteins...
Ben Wolozin: Definitely.
Inez Vincent: Based on the plascticity/neuronal activity idea, it might be toxic metabolites, oxidative damage, DNA damage, or other undetermined factors that may signal this special pathway in selective neurons.
Mark Smith: We have the classic chicken-and-egg situation: what causes what? We have tried oxidative stress, tau phosphorylation, and proteasome inhibition as initiators but do not see the appearance of all the other aspects. However, once neurons leave the G0 phase of the cell cycle, then we see all aspects appearing in a temporal progression.
Craig Atwood: Cell cycle re-entry also could explain the mitochondrial abnormalities (increased mtDNA and Cox1) seen in pyramidal neurons, and hence oxidative stress.
Rachael Neve: Mark, what model system are you using to look at temporal progression? Primary neuronal cultures?
Ping Lu: Can oncogenes be involved in activation of the cell cycle?
Karl Herrup: Ping, I think the answer is "not likely" since this would seem to necessitate an early genetic change and would be difficult to explain in terms of the large number of neurons that die. Cell Cycle and Cell Specificity
Inez Vincent: I wonder if it is possible that only certain neurons might be susceptible to using cell cycle-driven mechanisms in degeneration—for instance pyramidal neurons—whereas neurons of the substantia nigra, cerebellum, etc. do not?
Gabrielle Strobel: Inez, might that have to do with differences in activity/plasticity? How would you test that?
Inez Vincent: Gabrielle, possibly.
Robert Bowser: Inez, degenerative selectivity is probably related to plasticity, though cell cycle activation may only push the neuron towards a more susceptible state, whereby other factors (Aβ, etc.) may then ultimately lead to its demise.
Zsuzsanna Nagy: Bob, one wonders whether the neuronal populations that enter the cell cycle depend on personal susceptibility or maybe on the nature of the mitogenic stimulus…
Inez Vincent: Yes, Bob. However, cell cycle activation is prominent in certain other degenerative disorders, such as Niemann-Pick disease, that totally lack amyloid deposition.
Zsuzsanna Nagy: Inez, we found that granule cells with Pick bodies also follow the cell cycle path.
Craig Atwood: Inez, has not amyloid been shown to deposit with cholesterol in Niemann-Pick disease?
Nathaniel Milton: Inez, don't forget the glial cell cycle.
Karl Herrup: Let's not forget that the evidence for this cell cycle thing goes way back to the beginnings of the CNS. The breadth of applications to neurodegeneration is thus potentially much wider.
Ping Lu: I agree with Inez. It seems that neurons in certain regions are activated, but not others.
Zsuzsanna Nagy: Ping Lu, I think the neurons involved vary from one disease to another. Maybe individual sensitivities will determine which person gets what type of neurodegeneration.
Mark Smith: Inez, do you think that the initiating event is specific for certain neurons or that only pyramidal neurons develop the phenotype…?
Gabrielle Strobel: Current transgenic mice in AD (AβPP, PS, tau, ApoE) are all partial models. Have any of them been crossed with transgenic mice used in cell cycle and cancer research to see if that recapitulates AD more fully?
Mark Smith: Gabrielle, a nice thought, except that cancer involves successful division, this is something else.
Inez Vincent: Yes, Gabrielle, but it is impossible to ignore the selectivity. And here, activity and perhaps other biochemical features may be important, such as the amount of tau.
Douglas Feinstein: Inez, is there any correlation about which types of neurons are most affected, where they are, what transmitters they express?
Mark Smith: Douglas, I think people are 'gun shy' of this after the acetylcholine story...
Douglas: Right, Mark, but it might be nice to know. How about Cox2 survival versus cell cycle entry? Cell Cycle Interaction with Other Processes
Ping Lu: Pin1 may be a factor linking cell cycle, AD, and cancer.
Karl Herrup: Ping, perhaps you need to spell out your Pin1 idea a bit more for those of us who have just heard it here.
Ping Lu: Pin1 is critical for controlling G1/S and mitosis. It is required for dephosphorylation of many epitopes of the MPM-2 antibody. Pin1 is overexpressed in cancer but its function is inhibited in AD.
Nathaniel Milton: Mark, is the AD brain we all look at the survival success?
Mark Smith: To some extent yes, at least those neurons with pathology.
Rachael Neve: I'd like to propose that AβPP is intimately involved in this cell cycle process. We've shown that AβPP interacts with the cell cycle protein AβPP-BP1, and that activation of AβPP-BP1 can cause neurons to enter the cell cycle. Perhaps as we age, the interaction between AβPP and AβPP-BP1becomes unregulated. Due to oxidative stress or inflammation, maybe? Just to add to this, we also have evidence that NFkB activation inhibits AβPP-BP1’s ability to push neurons into the cell cycle, showing that NFkB is neuroprotective in this process.
Craig Atwood: Rachael, yes, AβPP-BP1 needs to be further explored.
Inez Vincent: Since Ping mentioned 'oncogenes' earlier, I wanted to draw everyone’s attention to the fact that the cdc2 gene lies in the new AD susceptibility region on chromosome 10!
Karl Herrup: Inez, I'd love the connection, but cdc2 is pretty late in the cell cycle, no?
June Kinoshita: Inez, that's interesting! Have you talked to any of the geneticists about this?
Nathaniel Milton: Gene differences in cdc2 have not been linked to AD yet. We’re still searching.
Inez Vincent: Great, Nathaniel!
Ping Lu: Plk1 is another oncogene involved in AD.
Karl Herrup: How is it involved?
Ping Lu: Plk1 is amplified and hyperactivated in cancer, it is also known to be activated in AD.
Ken Maiese: To add a little more fuel to the fire, especially with neuronal vulnerability, we have found that attempted cell-cycle regulation impacts both the early (membrane asymmetry) and late stages of neuronal injury (genomic DNA injury).
Rachael Neve: Ken, can you elaborate on that? Have you published it?
Ken Maiese: Yes, see Lin et al., 2001. We have shown that exposure to toxic injuries, such as free radicals, leads to progressive neuronal injury over 24 hours. This is accompanied by the rapid induction of the initial phase of programmed cell death that involves the exposure of membrane phosphatidylserine residues. Using the cell cycle markers bromodeoxyuridine (BrdU) and proliferating cell nuclear antigen (PCNA), we have illustrated that the progressive exposure of membrane phosphatidylserine residues occurs in concert with the attempt by post-mitotic neurons to enter the cell cycle. This ill-fated attempt occurs only in the same population of neurons that have begun the initial stages of programmed cell death, suggesting that attempted induction of a latent cell cycle in post-mitotic neurons correlates with the early stage.
Mark Smith: Far be it from me to defend the amyloidists, but let’s remember that neuronal injury/stress of any kind increases AβPP/Aβ, so it might be involved in so-called "non-amyloid" conditions.
Inez Vincent: That’s correct, Mark, and brings the presenilins into the fray as well!
Rachael Neve: Mark, to support your statement: A simple increase in AβPP levels causes neurons to enter the cell cycle. We've shown that in our primary neuronal culture model, where overexpression of AbPP by way of HSV vectors causes DNA synthesis.
Inez Vincent: Rachael, do you think that post-mitotic neurons would behave differently than primary neurons?
Rachael Neve: Inez, our primary cultures are largely postmitotic.
Karl Herrup: I still think a neuron that is driven into the cycle will die rather than divide. This implies that the specificity (at least to this type of death) must be in the signal, not in the neuron.
Inez Vincent: I agree, Karl.
Ping Lu: So do I, neurons will die if driven to divide. I think this is a major difference between cancer and AD.
Zsuzsanna Nagy: Rachael, did your neurons actually divide and, if so, what is the time required for cycle completion?
Rachael Neve: Our neurons don't divide; they die after showing DNA synthesis. We've shown that inhibitors of DNA synthesis don't prevent the death, but inhibitors of the G0-S phase transition do.
Robert Bowser: Zsuzsanna, the nature of the mitogenic stimulus will define the intracellular pathways activated and thus the presence of any cell cycle proteins activated.
Karl Herrup: Bob, I agree.
Inez Vincent: One thing we must then keep in mind is how this 'mitogenic' stimulus is propagated to different neurons and brain regions in AD.
Ken Maiese: We have found that a small percentage of neurons enter the cell cycle but do not succumb to injury. Any thoughts?
Robert Bowser: Ken, I think that a neuron’s susceptibility will depend on how far into (if at all) the cell cycle it truly progresses.
Karl Herrup: Ken, I think GABAergic neurons are spared this rule. Does anyone else find that GABAergic neurons in (cortical) cultures behave differently with regard to the cycle?
Inez Vincent: That’s interesting, Ken and Karl.
Rachael Neve: Is that right, Karl - do you find that GABAergic neurons aren't susceptible to cell cycle entry?
Karl Herrup: They are susceptible to cell cycle entry, just not to cycle-induced death.
Rachael Neve: That's very interesting. I assume you've determined that in a primary neuronal culture, using ICC coupled with BrdU labeling?
Karl Herrup: Yes, the results are from culture.
Inez Vincent: This would certainly need to be followed up with other neurotransmitter-specific lines.
Karl Herrup: I've been trying to find out from a number of pathologists whether GABA neurons are known to be specifically spared or targeted in AD. No answers so far.
Zsuzsanna Nagy: Karl, I don't think it has been looked at yet.
Ken Maiese: Actually, some believe that those neurons that enter the cell cycle but are not injured represent a "greater plasticity" and may function along the same lines as embryonic stem cells.
Karl Herrup: Ken, I think you are on to something. I wonder if these cells aren't the source of the neurospheres that everyone seems to culture out of adult brain.
Ken Maiese: Karl, that is a possibility. Many questions still exist. We also know that in neuronal models, attempted cell cycle regulation accounts for approx 40-50 percent of the neurons injured, so other more downstream cellular pathways also play a role.
Yuzhi Chen: Dr. Herrup, I like your idea about signaling and neuronal death, and it may be through AbPP.
Cell Cycle Models of Disease States
June Kinoshita: Are there any mouse models in which cell cycle regulators are expressed in postmitotic neurons.
Ping Lu: June, we may have a mouse model.
Rachael Neve: Pray tell, Ping!
June Kinoshita: Yes!
Zsuzsanna Nagy: What have you got, Ping?
Ping Lu: The pin1-related mouse model.
Rachael Neve: What do you mean by that?
Inez Vincent: In Niemann-Pick disease type C, we have found that Purkinje neurons contain hyperphosphorylated tau and mitotic proteins. These cells die very early in the disease, but never form NFT. I believe a similar mechanism operates in them, and they die but without forming NFT. (paper under review).
Craig Atwood: Inez, for amyloid in Niemann-Pick Type C cells, see Yamazaki et al., 2001
Karl Herrup: Inez, have you looked at Ataxia-telangiectasia?
Inez Vincent: I'd love to look at ATM, but no time! But June, producing a cell cycle model in brain is going to be difficult, and may have to be approached sort of wildly!
Robert Bowser: June, I have looked at changes in retinoblastoma and E2F1 in AβPP and SOD1 transgenic mice. We haven't seen any changes in the AβPP transgenic mice, and little neuronal cell death occurs in these animals. However, we did see changes in the SOD1 mice prior to the onset of motor neuron cell death.
Ken Maiese: Bob, I definitely agree with you. We find, though, that the cells appear to stop prior to G1, similar to several other published reports.
June Kinoshita: Bob, just to be clear, you did see cell cycle gene expression in SOD1 mice prior to neuronal degeneration?
Robert Bowser: June, yes, prior to and concurrent with mitochondrial vacuolation. We are currently following up these observations and hope to better define the temporal pattern and relationships (if any) to cell death.
Inez Vincent: Some of you may hate me for saying so, but it really is not possible to use neuronal cultures (even if they are 'differentiated') as models for cell cycle-mediated degeneration per se. Primary neurons and neuronal cell lines are sustainable in cell culture primarily because of their cell cycle abnormalities. It would be far too difficult to interpret results obtained with these cultures in the context of cell cycle mediated events in mature, differentiated neurons of brain. This does not mean that neuronal cultures are not useful for studying many other aspects of neuronal death, for example cytoskeletal abnormalities, oxidative damage pathways, and other signaling pathways.
Zsuzsanna Nagy: I agree with Inez there, unfortunately.
Glenda Bishop: Inez, if neuronal cultures are not good models, what would be?
Karl Herrup: Inez, I think the problem is not with the culture per se but with the fact that we use embryonic neurons as the source of our cultures.
Nathaniel Milton: Inez, we need the right mouse model for AD. AβPP transgenics aren't perfect.
Karl Herrup: If we could culture adult, isolated neurons we might make new progress.
Inez Vincent: I agree with Nathaniel and Karl.
Ken Maiese: Inez, I agree all models have their drawbacks, but we use primary cultures, not cell lines. Obviously, the knowledge gathered from cultures must be translated into animal models.
Rachael Neve: I think neuronal culture models give a good first approximation, which can then be tested in vivo.
Ping Lu: I think mouse models will be a better choice.
Gabrielle Strobel: Towards better in vivo models: Would it be useful to modify expression of various cell cycle genes in tau-transgenic drosophila to make a more faithful model of neurodegeneration? Or create other drosophila models?
Zsuzsanna Nagy: I don't like the drosophila idea.
Gabrielle Strobel: Why not?
Zsuzsanna Nagy: I think it is a bit far away from what we are trying to find. Call me old-fashioned!
Mark Smith: Greg Brewer has a technique to culture adult neurons. He claims even to get them to divide...the data/video are spectacular! ( Brewer, 1999.)
Zsuzsanna Nagy: Karl, what about the Brewer method? Have you tried it?
Karl Herrup: We have tried the Brewer technique without success. I’ve seen the video and wonder if these are the GABA neurons.
Zsuzsanna Nagy: We didn't have much luck with the method either. That's why I asked.
Inez Vincent: The Brewer technique works but only because the cells are not truly post-mitotic. How then, can one study cell cycle-associated mechanisms in such cells? The real challenge is turning on the cell cycle in a real post-mitotic neuron in vivo!
Nathaniel Milton: A leading journal editor suggested I use cdc2 knockouts - any better ideas?
Ping Lu: A cdc2 knockout is likely lethal?
Gabrielle Strobel: I noticed an interesting letter by David Smith, Zsuzsanna, and others reporting that cyclin E is upregulated in hippocampus in people with elevated homocysteine and AD. Any thoughts?
Inez Vincent: This is a great study. I hope there will be a follow up?
Zsuzsanna Nagy: I am working on it...
Karl Herrup: Gabrielle, Mark and I talk about the difference between a toxic response and a protective one. Both will come up in a disease state but you only want to inhibit the latter.
Mark Smith: Inez, I think there is a big difference between a conventional cell cycle and a cell cycle in a post-mitotic neuron. To study AD, we should look only at the latter and therefore need post-mitotic cells.
Ping Lu: Mark, I agree that an AD cell cycle is different from the conventional cell cycle.
Zsuzsanna Nagy: I agree with Mark, but then we are back to the initial question: how to look at it?
Inez Vincent: The best, but also most difficult system to study is a post-mitotic neuron in brain.
Karl Herrup: Ping, Mark, I think the early cycle events are perfectly traditional, they just hit a road block and can't move on.
Mark Smith: If that's so then why do we see G1 markers simultaneously with S markers?
Ping Lu: I don’t think early cell cycle events are all normal.
Ken Maiese: Ping, I agree. It appears that induction of the cell cycle in neurons can lead to other events associated with caspase activation.
Inez Vincent: Our work supports Karl’s explanation. Neurons simply cannot get through mitosis.
Craig Atwood: Why the roadblock? Problems with cytokinesis?
Zsuzsanna Nagy: Craig, I think cytokinesis is the final block only.
Karl Herrup: Craig, DNA breaks are my guess (which might please the oxidation folks).
Gabrielle Strobel: Craig, or accumulated DNA damage and impaired ability to repair?
Physiological Role for Cell Cycle Proteins in Post-Mitotic Neurons
June Kinoshita: There's an assumption that the expression of cell cycle genes in postmitotic neurons is an ectopic event, but do we know that for sure? Has the possibility that they have some other function (perhaps at a lower level of expression) in postmitotic neurons been ruled out?
Rachael Neve: June, that's a very good point. I wonder if these cell cycle proteins that are in neurons aren't involved in normal neuronal plasticity. Quite a bit of evidence coming out to support that idea.
Ben Wolozin: Rachael, I agree. Understanding this is essential because it helps to determine whether one wants more cell cycle activation or less. The answer is not clear.
June Kinoshita: Rachael caught the subtext of my question. Any thoughts from others?
Mark Smith: June, always a possibility since cell cycle proteins have other functions. Not clear.
Karl Herrup: June, I've not heard of any use for mitotic events in post-mitotic neurons. But I could be persuaded.
Rachael Neve: Karl, for example, there are data showing that there is increased neurogenesis with learning; also, some forms of plasticity such as ischemia and drug abuse cause changes in cell cycle proteins.
Karl Herrup: True Rachael, but the increased neurogenesis is in a normally dividing stem cell population. I don't know the drug abuse data.
Rachael Neve: The drug abuse data are out of Eric Nestler's lab.
Inez Vincent: My response to June’s comment is that we need to launch a serious study of cell cycle proteins in normal human brain so we better understand the picture in a normal neuron. These proteins have not been previously studied in brain, and so we know nothing about what they might be doing in normal neurons.
Rachael Neve: I agree.
Jan: Perhaps the roadblock to actual mitosis is that activation of cell cycle proteins does not occur in a coordinated fashion.
Mark Smith: Agreed.
Nathaniel Milton: Nuclear versus cytoplasmic actions of cell cycle kinases need to be assessed.
Rachael Neve: And actions at the synaptic terminal, as well!
Robert Bowser: Cell cycle proteins in the nucleus likely participate in regulating not only gene expression but also chromatin structure. Therefore changes in nuclear cell cycle proteins could also modify chromatin structure and make neurons more susceptible to DNA injury.
Karl Herrup: Nathanial and Bob, I think this is getting down to it. A lot may ride on localization of these compounds.
Mark Smith: Bob, chromatin remodeling is certainly an issue that we are looking at closely.
Gabrielle Strobel: But aren't many of the observed cell cycle proteins in the cytoplasm, not in the nucleus?
Inez Vincent: That’s correct, Gabrielle.
Karl Herrup: We find them there in postmortem material, but where were they in the pre-agonal state?
Nathaniel Milton: Phosphorylated Aβ and tau are cytoplasmic events.
Zsuzsanna Nagy: Exactly, it appears that while they relate to cell cycle events when localized in the nucleus, the cytoplasmic localization may actually be related to something else, such as tau phosphorylation.
Robert Bowser: Karl and Nathaniel, there are definitely changes in subcellular distribution of many of these proteins, some of which are not typical for cell cycle functions. So further defining function versus distribution is important to determine how these proteins function in protective versus degenerative pathways.
Gabrielle Strobel: Chromatin remodeling is also being looked at intensely in Huntington's.
Zsuzsanna Nagy: Gabrielle, yes, but I personally take extreme care to make sure we only look at nuclear localization in relation to cycling.
Rachael Neve: What do you mean by that, Zsuzsanna?
Zsuzsanna Nagy: I mean that cdks might drive cell division in the nucleus, but if they do not translocate there, they might go on phosphorylating whatever is at hand in the cytoplasm.
Craig: When a cell divides, what happens to the nuclear membrane?
Karl Herrup: Craig, in normal division the nuclear membrane dissolves in M phase.
Craig: Maybe it has dissolved earlier?
Inez Vincent: Craig, the nuclear membrane typically dissolves and reforms once cytokinesis is complete. There are no such signs of nuclear membrane changes in AD brain, even in neurons containing full-blown NFT.
Craig: Well, something must be wrong! Or are the data artifactual?
Nathaniel Milton: If tau and Aβ phosphorylation are behind AD pathology, we need chemicals which only block these events and not cell cycle, i.e. substrate specific-inhibitors.
Mark Smith: Nathaniel, only if tau and Aβ are BAD!
Nathaniel Milton: Phosphorylated Aβ ; is BAD, very very BAD.
Mark Smith: I continue to smile!
Nathaniel Milton: Mark, you obviously don't have any phosphorylated Aβ .
June Kinoshita: To elaborate on Ben's simple scenario [mentioned a while back], perhaps a neuron under stress is expressing cell cycle proteins to carry out some plasticity-related functions, but they build up because Aβ is gumming up the proteasomal system, and reach a level where they drive the neuron into mitosis?
Mark Smith: June, I respectfully disagree with your sequence of events.
Ping Lu: June, that is a good summary.
Karl Herrup: June, I'm not sure I can buy into this one completely. There still needs to be a trigger and a source of specificity.
Rachael Neve: I like your thought, June, all except for the Aβ part…
Inez Vincent: I'm with Rachael!
Ben Wolozin: Rachael, why don't you like the Aβ part?
Rachael Neve: Ben, you made me laugh out loud!
Nathaniel Milton: Aβ could be an effect, not the cause.
Zsuzsanna Nagy: Nathaniel, I like that one.
June Kinoshita: Well, I'm trying to mediate between all the camps. Everyone gets to play a role, but not everyone can be the star!
Gabrielle Strobel: Zsuzsanna, your diagnostic test based on G1/S control in lymphocytes, at what stage is that? Is a company developing it?
Zsuzsanna Nagy: Gabrielle, there is no definite answer to that one yet. Are you interested?
Gabrielle Strobel: I'll be interested in taking it in 20 years! In the test, are AD and control groups completely separate, or do they overlap somewhat?
Zsuzsanna Nagy: There is a small overlap. We are trying to figure that one out. It might have to do with the age-related changes we talked about earlier. To be useful, the test will have to have an age correction, somehow.
Rachael Neve: Which of the mouse models have been studied extensively for evidence of DNA synthesis? All of them? All without positive data?
Inez Vincent: I haven't seen any studies of DNA synthesis in vivoβvery difficult to do!
Rachael Neve: Karl's done it!
Nathaniel Milton: Zsuzsanna, but what is the cause? Cdc2 ?
Zsuzsanna Nagy: Nathaniel, It might be cdc2 or cdk2. That is what I believe, anyway.
Nathaniel Milton:Cdc2 is on chromosome 10 in the AD region. That’s my choice.
Zsuzsanna Nagy: Nathaniel, yes I heard Inez Vincent mention it before. I am looking forward to some reprints.
Inez Vincent: Karl mentioned earlier that it probably does not start with cdc2. At least in the cell cycle, gene expression is often coordinated. Whether this is true of neurons needs to be determined.
June Kinoshita: Zsuzsanna, you mentioned that cell cycle regulators are expressed in epileptic neurons. The mouse models for epilepsy should be pretty good. Has anyone looked at them?
Zsuzsanna Nagy: June, I think there were some studies but I haven't followed them closely.
Gabrielle Strobel: A basic question: Neuronal loss in AD starts in the entorhinal cortex and then proceeds in a fairly predictable anatomical pattern. Does the expression of cell-cycle markers match this pattern?
Inez Vincent: We looked at the brains of Huntington’s disease victims. Did not find any cell cycle aberrations. We have unfortunately not had access to the entorhinal region. If someone is willing to send me the tissue I would love to do that!
Mark Smith: Gabrielle, the cell cycle precedes in my opinion.
Gabrielle Strobel: Mark, I understand, but do cell cycle aberrations also proceed through the brain in the anatomical patterns seen for the neurodegeneration?
Zsuzsanna Nagy: Gabrielle, I don't know of a systematic study but in Down’s syndrome brains we find the cell cycle-related protein expression in areas where AD will develop for sure later.
Ken Maiese: Zsuzsanna, that is very interesting. Can you elaborate on the time frame of this?
Zsuzsanna Nagy: Ken, Downs’ patients develop AD when they are about 30-40 years. The cell cycle markers appear when they are in their teens.
Karl Herrup: Zsuzsanna, is this published? That's a great finding.
Ken Maiese: Zsuzsanna, how do you know that there exists a specific causal relationship. I realize, at this time, this is more of a hypothetical question, but as I mentioned before, we do not find that all cell cycle induction in neurons leads to injury.
Zsuzsanna Nagy: Ken, I think that depending on the age of the rat, you might find differences in neuron behavior and survival after mitogenic stimulation.
Inez Vincent: Zsuzsanna brings up a good point. The duration of the changes make it very difficult to study prospectively!
Zsuzsanna Nagy: Sorry Ken, I missed so much because I am a slow typist. What model did you use?
Ken Maiese: We use primary rat neurons.
June Kinoshita: Everyone, this has been very lively and informative. Can you propose some priority research questions to move this whole area of inquiry forward?
Karl Herrup: June, I think what we need are models. Culture or on the hoof. Right now our mice are letting us down when it comes to neuron death.
Ben Wolozin: I agree with Karl 300 percent (or $100,000 worth of agreement). One cannot study this issue in a causative, hypothesis-driven model unless there is a good animal model that recapitulates the cellular changes in AD. The P301L tau x Aβ model might be the best. Then one can ask what happens if the cell cycle changes are blocked.
Inez Vincent: I would like to suggest sharing tissues so a variety of different markers may be followed in the same cases and the results more meaningfully interpreted. I wish Steve Snyder were around to make an offer either of organizing this or paying for it!
Mark Smith: Inez, I can help you on this.
Inez Vincent: Awesome, Mark!
Nathaniel Milton: If we knockout the cell cycle inhibitors do we increase, reduce, or have no effect on neurodegeneration?
Karl Herrup: Nathaniel, good question
Ping Lu: Some cell cycle inhibitors have been knocked out. Some sections of brain are bigger (p27).
Inez Vincent: Next step: let’s get together to discuss a more comprehensive model for studying cell cycle changes in vivo. I have a few ideas and am working on some.
Gabrielle Strobel: As we have reached the end of the hours I want to thank everyone for participating before people start to drop out. Please do continue, though, for as long as you like.
June Kinoshita: Yes, thank you all very much for attending today's chat!
Karl Herrup: This was fun. I just wish I could type faster. Bye all.
Ping Lu: Thanks. It is a great discussion
Rachael Neve: Thanks! I really enjoyed this!
Mark Smith: June, seems from this and other discussions, that keeping up with references is getting harder and harder. Could you list references under subjects that people could download or otherwise use? This subject could be a great start.
June Kinoshita: Yes, in the transcript we dig up citations referred to in this discussion as best we can and also include all that you send us.
Ben Wolozin: I am signing out. Very interesting.
Zsuzsanna Nagy: Yes, it was good to 'see' you all. Bye
Robert Bowser: Bye all.
Craig Atwood: Great chat, bye.
Mark Smith: Thanks for a great chat..
Ken Maiese: Thank you for a great discussion. Take care everyone!!!
Inez Vincent: I had fun. June, Gabrielle, and Nico-thanks for taking the initiative in organizing this chat!
Background
Background Text
By Inez Vincent
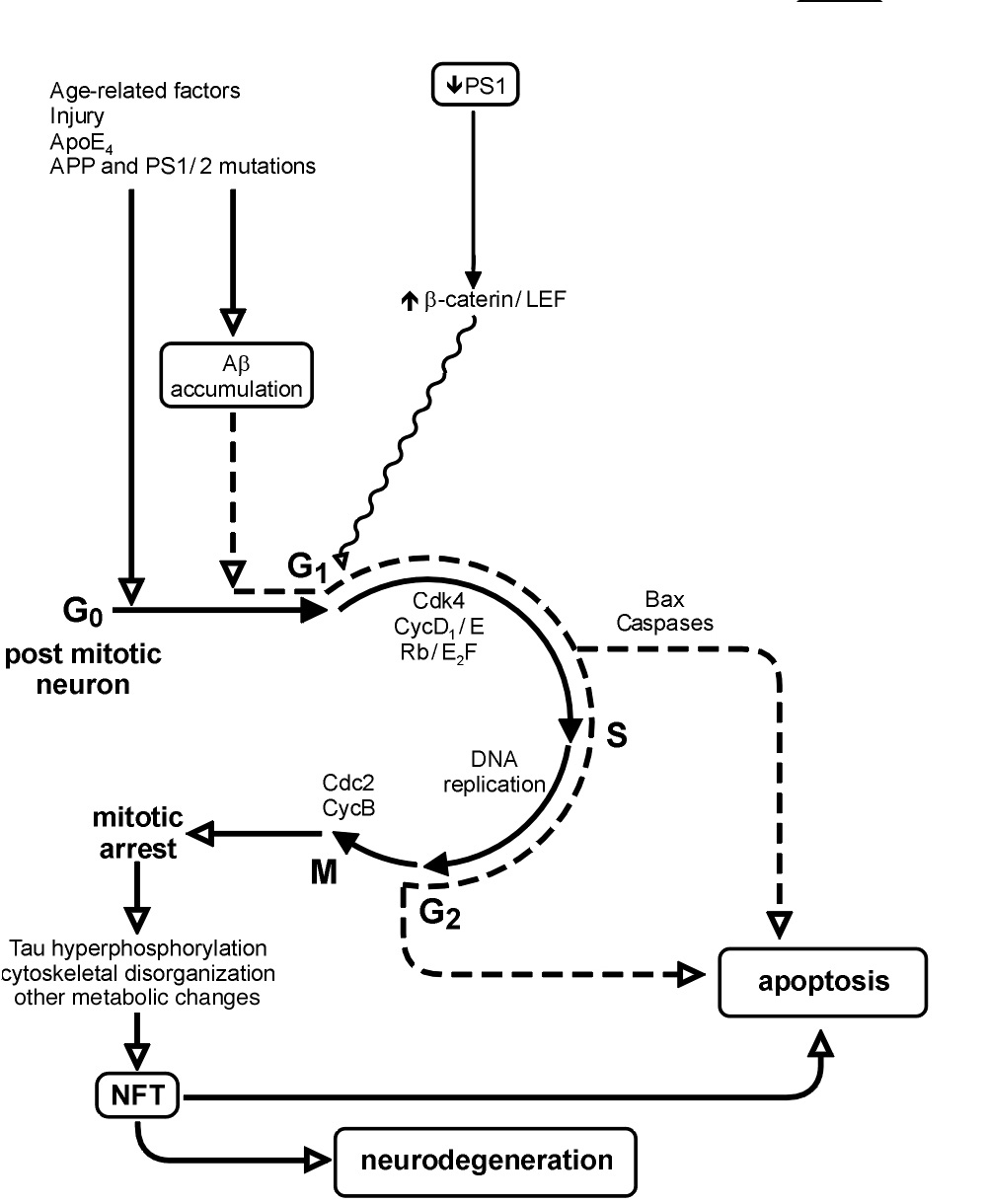
A fascinating mechanism for neurodegeneration in Alzheimer's disease has evolved in the last 5 years, namely that an inappropriate reactivation of the cell cycle is an early and important event in the development of AD. It defies all that we have believed about the terminal differentiation status of the neuron in mature brain. It implicates molecules that were thought to function exclusively as regulators of the cell division cycle and are not typically found in the largely non-proliferative brain.
This historic view was challenged recently on two fronts. First, studies of neurodegeneration in Alzheimer's and related disorders have indicated a resurgence of cell-cycle activity in degenerating, but not healthy, neurons. Second, techniques for detecting DNA synthesis in vivo and for unequivocal identification of newborn neuronal cells have demonstrated continued neurogenesis in normal adult brain. This newfound ability of adult brain to generate neurons from stem cells residing within the brain is being pursued intensely for its potential in replacing neurons lost to degeneration. However, we must realize that the neurogenic capacity of normal brain is quite distinct from the disease-associated reappearance of cell-cycle regulators, which takes place in neurons that have been postmitotic for decades since their origin in development.
Two major observations have helped establish the cell-cycle hypothesis for AD neurodegeneration. First, activation of cell-cycle regulators generally precedes formation of degenerative lesions, such as neurofibrillary tangles (NFTs), and the regulators and their downstream effectors eventually become incorporated into NFTs. Thus it has been postulated that cell-cycle regulators initiate and mediate the neurodegenerative process. Second, with the exception of karyo- and cytokinesis, markers from every phase of the cell cycle, and duplicated chromosomes, have been described in degenerating neurons. This has led to the hypothesis that vulnerable neurons re-enter the cell cycle and proceed through S phase, but then abort somatic division and eventually degenerate. I have been engrossed with this issue ever since our initial observations. Many questions preoccupy my mind:
- What triggers this 'final cycle,' and when?
- Are neurons truly postmitotic, or do they cycle very, very slowly?
- Is this unscheduled, inappropriate 'division' of postmitotic neurons akin to the dysregulated division of non-neuronal cells that culminates in neoplasia?
- Are neurodegeneration and neoplastic transformation - despite their opposite outcomes - driven by the same cellular mechanism?
- Are mutant AβPP, PS1 and PS2 neuro-oncogenes?
- Is neurodegenerative disease a 'cancer' of neurons?
A potential benefit of establishing such a connection is that it creates the opportunity for applying to the treatment of Alzheimer's disease a spectrum of candidate drugs being developed against cancer. Such a "joint venture" might provide a means for curbing two of the most costly health problems in our society.
If you are intrigued by this story, join us for our live online chat on May 20th, noon Eastern. To fuel our discussion, I have posited below some of the popular ideas pertaining to the cell-cycle hypothesis for AD neurodegeneration, along with data pro and con these ideas. Having chosen only a few sample references to support the arguments, I apologize to many who are not acknowledged. I am grateful to everyone who has contributed to establishing this new and exciting research area.
Postmitotic Neurons Degenerate Because of Inappropriate Cell Cycle Activation
| YES | NO |
| Cell cycle markers increased in affected neurons in AD. (Rev by McShea, 99; Zhu, 99; Nagy 00; Arendt, 00; see Neurobiol. Aging ,21, 00)
G0-G1: mitogenic/trophic factors and receptors, downstream signaling, increased pRb and E2F1 (Jordan-Sciutto, 02) G1-S: cdk4, cyclins E and A, PCNA, p105, Cdc25A, duplicated chromosomes. Mitotically phosphorylated proteins are incorporated into NFT. Pin1 is sequestered in NFT and depleted in neurons, promoting mitosis (Lu, 1999). Increased mitochondria. AβPP and tau undergo cell cycle-dependent phosphorylation in dividing cells (Suzuki, 94; Preuss, 98; Zhang 00). Forced expression of oncogenes in neurons, (Feddersen, 95) and other postmitotic cells (Crescenzi, 95), leads to cell cycle re-entry followed by death. |
Neuroscience dogma: terminally differentiated neurons incapable of division (Rakic, 85)
Increased cdk inhibitors p15, p16, p21, in AD (Arendt 96; McShea, 97) inconsistent with cdk activation. Cdks and their regulators restricted to neuronal cytoplasm, while DNA synthesis and mitotic initiation occurs in nucleus (Husseman, 00) No evidence for karyo- or cytokinesisSimultaneous increases in G1 and M of Cdk activity is inconsistent with exquisite control of temporal fluctuations in normally dividing cells. Chromosomal reduplication may simply increase with aging (Goldberg, 84; Medvedev, 86; Borsatto 98; Fujisawa, 98; Wagner 01).Aneuploidy in neurons of mature mouse brain (Rehen, 01), has never been studied in human brain |
What We Need: In vivo models to establish the role of cell-cycle regulators and progression of each cell-cycle phase in neurodegeneration.
Technical Caveat: Differentiated neuroblastoma and primary neuron cultures are quick and easy, but are not sustainable without cell-cycle aberrations. Neuroblastoma are transformed cells, often with gross chromosomal and checkpoint abnormalities. Primary neurons do not down-regulate cell cycle genes until 4-5 days in culture but most transfection experiments are initiated before this time. Primary neurons do not down-regulate cyclin B1 expression until about 10 days in culture! These systems are not truly postmitotic. They are OK for one-step relationships (e.g. does ectopic cdc2 expression generate mitotic phosphoepitopes,) but not for unraveling cell cycle mechanisms, signaling pathways, cellular phenomena.
In AD, Non-Neuronal Cells Also Have a Cell Cycle Defect
| YES | NO |
| Failure of proliferation control in blood lymphocytes, low responsiveness to mitogenic compounds (Steiler, 01), higher sister chromatid exchange compared with young adults (Melaragno, 91), decreased responsiveness of lymphocytes to G1 inhibitors (Nagy, 02), increased proliferation of lymphoblasts (Urcelay, 01) | No changes in T lymphocyte subsets or proliferation (Leffell, 85), no change in mitogenic response of lymphocytes to phytohemagglutinin, pokeweed mitogen, concavalin A, and staph protein A (Araga, 90) |
The Known Etiologic Factors AβPP, PS1/2 Are Consistent With the Hypothesis
| YES | NO |
| Extracellular AβPP enhances proliferation of CNS-derived neural stem cells (Hayashi, 94; Ohsawa, 99), induces mitosis of Schwann cells (Alvarez, 95) is trophic for fibroblasts, epithelial (Pietrzik, 97) and epidermal basal cells (Hoffmann, 00), lymphocyte proliferation (Trieb, 96), PC12, neurons (Yamamoto, 94), Intracellular AβPP in colon carcinoma cells (Meng, 01). Overexpression of wild-type AβPP and FAD mutant AbPP-induced DNA synthesis in primary cortical neurons (Neve, 00) mediated by AβPP-BP1.Aβ induces cell cycle signaling and neuronal death (Copani, 99; Giovanni, 0l ;Wu, 00).
PS-1 promotes neurogenesis in Xenopus (Paganelli, 01); PS1 deficiency increases cyclin D1 and entry into S phase, reversed by PS1 reexpression, but not by FAD mutants (Soriano, 01), PS-1 deficiency in cancer cells leads to tumor suppression, suggesting that PS-1 is required for tumor formation (Roperch, 98). 2 FAD mutations predispose to chromosome missegregation (nondisjunction) as evidenced by increased chromosome 21 trisomy mosaicism (Geller, 99), and an increase in association of a PS-1 intron 8 polymorphism in mothers of Down syndrome patients with meiosis errors (Petersen, 00); cdc2 binds and phosphorylates Aβ (Milton, 01). Cdk inhibitor blocks neuronal apoptosis in AbPPswe/ PS1-A246E mice (Xiang, 02) |
No in vivo evidence exists that AbPP or FAD mutations causes cell cycle activation in neurons.Aβ inhibits endothelial cell replication in vitro (Grammas, 95).
Overexpression of PS-1, PS-2, in HeLa cause G1 arrest in HEK2293 cells in S phase (Jeong, 00), and FAD mutants potentiate arrest (Janicki, 00) - does not affect levels of p21. No evidence for direct effects of PS-1 or 2 on cell cycle activation in mature brain.Brain PS-1 expression is downregulated in p53 deficient mice (Amson, 00). |
What We Need: Examine cell cycle markers in the brains of mutant AβPP, PS, and AβPP/PS double transgenic mice; study effects of AβPP, PS, and ApoE4 on expression of specific cell cycle genes in differentiated primary neurons or neuroblastoma cells.
Cell Cycle Regulators Mediate Apoptosis, Not Cell Cycle Entry/Progression
| YES | NO |
| (Rev Cotman, 00; Shimohama, 00; Yuan 00)
DNA damage and fragmentation is increased in AD neurons, morphological apoptotic changes evident (Andersen, 01; Broe, 01). Cdk4 is an obligatory mediator of Bax- and caspase3-driven neuronal apoptosis, and could trigger apoptosis Caspases 1, 2, 3, 6, 8, 9, and 12 are implicated in Aβ-induced neuronal death in vitro, in animal models of neurodegen. diseases, and in AD brain (Roth, 01). Caspase cleavage products fodrin (Cotman, Rohn, 01), Bax, ZIP kinase, Bim/BOD, Bcl-2 and p21 (Yuan, 00; Engidawork, 01)are increased. 14-3-3 (Fountoulakis, 99), G1 cdks and cyclins are activated in in-vitro and in-vivo models for neuronal apoptosis, (Freeman, 94; Padmanabhan, 99; Liu, 01; Osuga, 00; Katchanov, 01; Chopp, 01) |
(rev Raina 01; Roth 01)
Frequency of DNA fragmentation in neurons too high to account for continuous, slow, neuronal loss over protracted disease period (Perry, 98) Increased cell cycle activity and NFT in neurons with intact nuclear membrane, no chromosomal condensation, no blebbing (Bancher 97; Husseman, 00; Raina 01) MAP kinase-phosphorylated c-Myc does not colocalize with caspase-3 activation in AD (Ferrer, 2001). |
What We Need: Double labeling of AD brain sections with cell cycle and apoptotic markers to determine temporal and spatial overlap, analyses of cell cycle markers in transgenic mouse models displaying apoptosis
Activation of Cell Cycle Regulators Is a Regenerative Response
| YES | NO |
| Increased Neuritic sprouting in AD. Extracts from AD brain stimulate branching of neurites in PC12 cells (Kittur, 92).
Increased embryonic a-tubulin mRNA in AD (Miller 90).Increased 'fetal' tau phosphoepitopes in AD (Hasegawa, 93; Bramblett, 93; Goedert, 93). Increased levels of growth-associated proteins GAP-43 (Martzen 93; de la Monte, 95), spectrin, (Masliah, 91), MARK (Drewes, 98) in AD. Brain injury can promote neurogenesis:Increased cyclin D, cdk4, neurogenesis in response to ischemia (Chopp, 01, Osuga, 00; Jiang, 01; Kernie, 01), andincreased NSE and NF-positive mitotic figures after partial cortical ablation in adult rat (Huang, 90) |
No evidence supporting a role for cell cycle regulators in regenerative neuritic sprouting in AD or any model.
No evidence for activation of cell cycle mechanisms in postmitotic neurons following injury |
What We Need: Double labeling for cell cycle markers and sprouting markers to illustrate presence in same neurons.
Cell Cycle Regulators Have Diversified Roles in Neurons
| YES | NO |
| Cdc25A and B, and Wee1 are constitutively active in normal postmitotic neurons of brain (Ding 00; Tomashevski 01; Vincent 01).
Cyclins A and B1 are commonly expressed in postmitotic neurons of middle aged and elderly humans (Pae and Vincent, unpublished), as are cyclin H (Jin, 99), andanaphase-promoting complex (Gieffers, 99). |
Expression of cell cycle genes is generally not detected by whole brain Northern blot or PCR analysis. (However, the brain is not a homogeneous organ. If expression is localized to specific neuronal populations or brain regions, it may be missed by these methods) |
What We Need: Systematic demonstration of such molecules by immunohistochemistry, western blot, in situ hybridization, PCR, in different regions of autopsy and biopsy brain from human/other primates; direct demonstration of the functions of these proteins in postmitotic neurons in vitro and in vivo.
Comment by Thomas Arendt
How do Cycling Neurons and AD Fit into the Big Scheme of Things?—Posted 14 May 2002
We understand a thing if we understand how it develops. This applies to both Alzheimer´s disease and the human brain.
Alzheimer´s disease is a chronic disorder with progressive neurodegeneration associated with a typical pathology: amyloid deposits and neurofibrillary tangle formation. While amyloid pathology is more uniformly distributed throughout the brain, tangle formation in different brain areas follows a particular sequence and always affects some areas earlier than others. Understanding this selective neuronal vulnerability to tangle formation is the key issue to understand Alzheimer´s disease.
Those brain areas and neuronal types that are highly vulnerable to tangle formation also differ from the rest of the brain in several other regards that might give us hints: They exhibit a particularly high degree of synaptic plasticity (and probably synaptic turnover), they have been acquired late during phylogenetic development (or have been completely re-organized during recent phylogenetic development), and they mature rather late during ontogenetic development.
Neurons are different from most other cells in the body in that they are highly polar and, thus, are probably the most differentiated cells in the true sense of the word. After proliferation, migration, and differentiation, they become integrated for a lifetime in a neuronal network. This integration requires intercellular communication that is largely regulated through synaptic plasticity. The neuron runs into problems, as cell connectivity and attachment are mechanisms that, during evolution from single-to multicellular systems, were acquired to control proliferation, differentiation, and cell death. For the differentiated, non-proliferative neuron, however, these mechanisms have become part of its genuine, plastic function. It is a common principle of evolution to re-use, at a certain phylogenetic point, a regulatory mechanism for a new biological function. For the neuron, controlling its synaptic plasticity is a great achievement, yet to do so at the expense of differentiation control may also put it at a permanent risk. Those neurons acquired late during brain evolution, for example cortical associative circuits that subserve typically human, higher cortical functions including learning, memory, reasoning, consciousness etc, need a particularly high degree of synaptic plasticity. This makes them supremely sensitive to lose differentiation control (and die?). While this appears like an evolutionary dead end, there is no selective pressure on this dead end as diseases associated with it, such as Alzheimer´s, occur after the reproductive period has ended.
Understanding Alzheimer´s disease and neurodegeneration within the framework of cellular differentiation and cell division control is an old idea. It first came up early in the 20th century (Cajal, 1928; Bouman´s theory of 'hyperdifferentiation,' 1934) and was repeatedly emphasized afterwards. Neurotrophic agents were suspected of exacerbating the pathologic cascade of Alzheimer´s disease, including its "aberrant neuronal growth". (Butcher and Woolf, 1989; Arendt, 1993; Heintz, 1993). It then became increasingly clear that mitogenic pathways in neurons are activated during early Alzheimer´s disease (Saitoh et al., 1993; Gärtner et al., 1995). More recently, insight into cell cycle control has seen major advances, been awarded a Nobel Prize, and the cell cycle itself came into direct focus of Alzheimer Research.
And still, we do not know how cellular differentiation is controlled in a neuron. Clearly, Alzheimer research might gain much if it learned from cancer biology and especially from developmental neurobiology.
I suggest these critical issues need to be tackled:
- How is neuronal differentiation regulated, and how is the differentiated stage fixed for a lifetime?
- What is the relationship between synaptic plasticity and neuronal differentiation? How do neuronal attachment, (synaptic) connectivity, and mitogenic stimulation come into these processes?
- Mechanisms known to mediate mitogenic effects during development: What are they doing in the adult brain (e.g. activation of the p21ras/MAPK pathway)?
- Is it possible to stabilize a neuron in its differentiated stage? Is this neuroprotective? Is this bad for synaptic plasticity and, thus, a restriction for higher brain function? Thomas Arendt, University of Leipzig, Germany.
References:
Ramon y Cajal S., 1928. Degeneration and regeneration of the nervous system. Oxford University Press, London.
Bouman L., 1934. Senile plaques. Brain 57, 128-142.
Arendt T, 1993. Neuronal dedifferentiation and degeneration in Alzheimer's disease. Biol. Chem. Hoppe-Seyler 374, 911-912.
References
Webinar Citations
Other Citations
{kind=link}
External Citations
Further Reading
Papers
- Joseph JA, Shukitt-Hale B, Denisova NA, Prior RL, Cao G, Martin A, Taglialatela G, Bickford PC. Long-term dietary strawberry, spinach, or vitamin E supplementation retards the onset of age-related neuronal signal-transduction and cognitive behavioral deficits. J Neurosci. 1998 Oct 1;18(19):8047-55. PubMed.
- Mosch B, Morawski M, Mittag A, Lenz D, Tarnok A, Arendt T. Aneuploidy and DNA replication in the normal human brain and Alzheimer's disease. J Neurosci. 2007 Jun 27;27(26):6859-67. PubMed.
Comments
Reply from Inez Vincent
Definitely, this would be worthwhile. Recent data supports the idea that phosphorylation of APP at the thr668 site alters its interaction with Fe65, and beta-amyloid production (Ando, 2001).
What needs to be determined is which kinase(s) phosphorylate(s) this site in vivo, especially in AD. Present evidence has implicated cdk5 (Iijima, 2000), cdc2 (Suzuki, 1994; Milton, 2001 and 2002), GSK-3β (Aplin, 1996), stress-activated protein kinase 1b (Jun N-terminal kinase-3) (Standen, 2001) and a novel kinase (Isohara, 1999).
One way to approach this problem is to inhibit specific kinases in amyloid producing transgenic mice and determine whether this has any effect on amyloid production, and another would be to examine APP processing in many of the transgenic mouse models over expressing one of the proline-directed kinases, or mice deficient in activity of one of them.
References:
Ando K, Iijima KI, Elliott JI, Kirino Y, Suzuki T. Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of beta-amyloid. J Biol Chem. 2001 Oct 26;276(43):40353-61. PubMed.
Iijima K, Ando K, Takeda S, Satoh Y, Seki T, Itohara S, Greengard P, Kirino Y, Nairn AC, Suzuki T. Neuron-specific phosphorylation of Alzheimer's beta-amyloid precursor protein by cyclin-dependent kinase 5. J Neurochem. 2000 Sep;75(3):1085-91. PubMed.
Suzuki T, Oishi M, Marshak DR, Czernik AJ, Nairn AC, Greengard P. Cell cycle-dependent regulation of the phosphorylation and metabolism of the Alzheimer amyloid precursor protein. EMBO J. 1994 Mar 1;13(5):1114-22. PubMed.
Milton NG. Phosphorylation of amyloid-beta at the serine 26 residue by human cdc2 kinase. Neuroreport. 2001 Dec 4;12(17):3839-44. PubMed.
Milton NG. The amyloid-beta peptide binds to cyclin B1 and increases human cyclin-dependent kinase-1 activity. Neurosci Lett. 2002 Apr 5;322(2):131-3. PubMed.
Aplin AE, Gibb GM, Jacobsen JS, Gallo JM, Anderton BH. In vitro phosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase-3beta. J Neurochem. 1996 Aug;67(2):699-707. PubMed.
Standen CL, Brownlees J, Grierson AJ, Kesavapany S, Lau KF, McLoughlin DM, Miller CC. Phosphorylation of thr(668) in the cytoplasmic domain of the Alzheimer's disease amyloid precursor protein by stress-activated protein kinase 1b (Jun N-terminal kinase-3). J Neurochem. 2001 Jan;76(1):316-20. PubMed.
Isohara T, Horiuchi A, Watanabe T, Ando K, Czernik AJ, Uno I, Greengard P, Nairn AC, Suzuki T. Phosphorylation of the cytoplasmic domain of Alzheimer's beta-amyloid precursor protein at Ser655 by a novel protein kinase. Biochem Biophys Res Commun. 1999 May 10;258(2):300-5. PubMed.
Ohio State University
I wanted to join this the very interesting discussion but was unable to do so. As researcher at ALS-Therapy Development Foundation, I am involved in discovering new drugs to treat ALS. I propose a model that may explain the transition of neurons into the cell cycle and their eventual death. It goes like this:
1. Proteasomal dysfunction affects degradation of various cell-cycle regulators.
2. We believe polyamines are among the main players in this cell-cycle dysregulation.
3. Ornithine decarboxylase (ODC), the enzyme that converts ornithine to the polyamine precursor putrescine, is regulated at the post-translational level by the proteasome.
4. The polyamine-inducible protein antizyme and ODC interact, and proteasomal inhibition alters the proteasomal breakdown of ODC by antizyme.
5. This can lead to ODC accumulation and high polyamine levels in cells.
6. High polyamine levels can signal cells to enter the cell cycle by chromatin and histone destabilization and other mechanisms.
7. The conflicting proliferation and inhibitory signal can drive cells into apoptosis/death. - Tennore Ramesh, ALS Therapy Development Foundation, Newton, Massachusetts.
References:
Toth C, Coffino P. Regulated degradation of yeast ornithine decarboxylase. J Biol Chem. 1999 Sep 3;274(36):25921-6. PubMed.
Chattopadhyay MK, Murakami Y, Matsufuji S. Antizyme regulates the degradation of ornithine decarboxylase in fission yeast Schizosaccharomyces pombe. Study in the spe2 knockout strains. J Biol Chem. 2001 Jun 15;276(24):21235-41. Epub 2001 Mar 30 PubMed.
Virgili M, Necchi D, Scherini E, Contestabile A. Increase of the ornithine decarboxylase/polyamine system and transglutaminase upregulation in the spinal cord of aged rats. Neurosci Lett. 2001 Aug 17;309(1):62-6. PubMed.
Bernstein HG, Müller M. The cellular localization of the L-ornithine decarboxylase/polyamine system in normal and diseased central nervous systems. Prog Neurobiol. 1999 Apr;57(5):485-505. PubMed.
Bernstein HG, Müller M. Increased immunostaining for L-ornithine decarboxylase occurs in neocortical neurons of Alzheimer's disease patients. Neurosci Lett. 1995 Feb 17;186(2-3):123-6. PubMed.
Salzberg A, Golden K, Bodmer R, Bellen HJ. gutfeeling, a Drosophila gene encoding an antizyme-like protein, is required for late differentiation of neurons and muscles. Genetics. 1996 Sep;144(1):183-96. PubMed.
Banner Research Institute
Inez Vincent's background text is excellent. I would make one detailed comment about the part where she refers to Martzen et al., 1993, as showing an increase in GAP-43. In this paper we showed what we presumed to be a move of phospho GAP-43 between a cytosolic compartment and a membranous compartment. In fact, other papers from my lab have shown decreased expression of GAP-43 in AD homogenates of frontal association cortex (e.g. Coleman et al., 1992). Subsequently we showed that GAP-43 message was decreased in NFT neurons relative to adjacent NFT-free neurons (Callahan et al., 1994), suggesting that the decrease we saw in homogenates was largely (not wholly - see next paragraph) due to those neurons with NFT rather than an equivalent decrease in all neurons. We cannot ignore the fact that papers exist in which immunohistochemical evidence indicated local GAP-43 immunoreactive sprouting. I believe these data and ours may be compatible if one assumes that although there may be some local sprouting, when looked at more globally in affected regions there is a net loss of GAP-43 expression. In one of our papers, we also showed no change in expression of GAP-43 in cerebellum in AD (Cheetham et al., 1996).
A paper in press in Neuropath Exptl Neurol., using double immunohistochemistry (for NFT and for several phospho tau sites as well as in-situ hybridization for synaptophysin message, now shows convincingly that synaptophysin message decreases depending on phospho tau immunoreactivity. Phospho serine 396/404 does not seem to affect synaptophysin message expression in the cell body, but phospho serine 262 does (both the former occur in the absence of frank NFT). Formation of frank NFT results in a further reduction of synaptophysin message expression. Incidentally, these single neuron data were almost precisely the same regardless of whether the sections sampled came from Braak 5-6 or Braak 2-3 AD, suggesting that a tangle neuron is a tangle neuron and a phospho-tau neuron is a phospho-tau neuron without regard to disease state. In other words, within neuron types (e.g. pyramidal, stellate, etc., and regional location) cells with similar immunohistochemically-defined phenotypes are similar with respect to other variables, too, regardless of the disease stage of the brain. What makes the difference in overall disease status is the number of neurons in state X.
References:
Martzen MR, Nagy A, Coleman PD, Zwiers H. Altered phosphorylation of growth-associated protein B50/GAP-43 in Alzheimer disease with high neurofibrillary tangle density. Proc Natl Acad Sci U S A. 1993 Dec 1;90(23):11187-91. PubMed.
Coleman PD, Kazee AM, Lapham L, Eskin T, Rogers K. Reduced GAP-43 message levels are associated with increased neurofibrillary tangle density in the frontal association cortex (area 9) in Alzheimer's disease. Neurobiol Aging. 1992 Nov-Dec;13(6):631-9. PubMed.
Callahan LM, Selski DJ, Martzen MR, Cheetham JE, Coleman PD. Preliminary evidence: decreased GAP-43 message in tangle-bearing neurons relative to adjacent tangle-free neurons in Alzheimer's disease parahippocampal gyrus. Neurobiol Aging. 1994 May-Jun;15(3):381-6. PubMed.
Cheetham JE, Martzen MR, Kazee AM, Coleman PD. Gap-43 message levels in anterior cerebellum in Alzheimer's disease. Brain Res Mol Brain Res. 1996 Feb;36(1):145-51. PubMed.
Ohio State University
Reply by Tennore Ramesh
I agree with Dr. Wolozin. Many cell cycle proteins are regulated by the
proteasome and that may be a simple starting point. CDk5 is shown to be
altered in ALS. P35, the neuron-specific activator of CDK5, is regulated by
the ubiquitin-proteasomal pathway and its half life is prolonged by
proteasomal inhibition. Other pathways involved in cell cycle, such as the
Jak-Stat pathway, are also regulated by the proteasome. Jak3 kinase
inhibition significantly extended the life of SOD1 G93A mice, suggesting
that modulation of these pathways may be a viable strategy in ALS and other
neurodegenerative diseases.
Leeds Trinity University
The Cdc2 kinase phosphorylates amyloid-beta (Milton NGN.
NeuroReport 12, 3839-3844, 2001) suggesting that cell cycle abnormalities may
influence the plaque formation and neurodegeneration associated with elevated
amyloid-beta. The amyloid-beta peptide also activates cdc2 directly and binds
to cyclin B1(Milton NGN. Neurosci. Lett. 322, 131-133, 2002). Perhaps it is
time to look at phosphorylated amyloid-beta as a causative agent rather than
the normal unphosphorylated form. This form may also provide a more useful
marker for neurodegenerative changes.
Boston University School of Medicine
Reply by Benjamin Wolozin
I was pleased to see Dr. Ramesh's comment on the importance of the
proteasome, and want to reiterate that proteasomal inhibition provides a
simple explanation for the cell cycle connection to AD. Proteasomes
regulate many cell cycle proteins, which certainly includes ODC, however I
don't think that there is compelling evidence to suggest whether there is a
particular cell cycle protein that triggers the entire process observed in
AD. Thus, ODC is likely to be regulated by the proteasome, but whether it
is the central protein in required for activating the cell cycle in AD brain
remains unclear.
Make a Comment
To make a comment you must login or register.