Does LATE Subvert Alzheimer's Trials? Biomarkers, Please!
Quick Links
Limbic predominant age-related TDP-43 encephalopathy, aka LATE, appears to dampen an aging person's memory in a similar way as does Alzheimer’s disease. TDP-43 inclusions occur as the lone pathology in some people, and alongside the plaques and tangles of AD in others. Alas, there are no biomarkers for TDP-43 inclusions. This poses a tricky challenge for the diagnosis and management of both disorders. At LATE 2022, a virtual meeting hosted by National Institute on Aging on February 11, trialists voiced concern that undetected LATE neuropathology in some enrollees of AD drug trials might skew the trial’s outcomes. They also said the field needs trials specifically for LATE.
- LATE neuropathology might mess with outcomes in some AD trials.
- Scientists need biomarkers to know it’s present in a person’s brain.
- Exosomes in plasma are a promising prospect.
- Structural MRI and FDG-PET may also detect LATE.
Researchers described their quest to find biomarkers for LATE. Soluble TDP-43 packaged within astrocytic exosomes detected in blood tracked remarkably well with LATE pathology. PET tracers for the TDP-43 inclusions remain elusive, but other neuroimaging modalities, for example structural change in the hippocampus and patterns of hypometabolism, may help identify people with LATE. Even so, scientist at the meeting agreed that much more work needs to be done to characterize these early leads.
LATE and Trials
Given recent work tying LATE to cognitive decline (see Part 2 of this series), scientists now think it’s quite plausible that this limbic pathology, unbeknownst to trialists, could influence outcomes in AD clinical trials. For example, LATE could mask otherwise positive results of drugs targeting plaques or tangles.
Reisa Sperling, Brigham and Women’s Hospital, Boston, leads secondary prevention trials for AD. She wondered to what extent LATE might explain outliers in those studies—i.e., people who decline much faster than was predicted based on their levels of plaque and tangle pathology at baseline. To get a sense of this, Sperling looked to observational studies, including the Harvard Brain Aging study, ROS-MAP, and ADNI. She noted that while as a group, cognitively normal people with amyloid plaques are likely to slip on cognitive tests in the coming five years, individual trajectories vary dramatically.
Similarly, tau tangles spread at varying rates in the brain. While they eventually invade the neocortex in most people with elevated plaque burden—a scenario Sperling and others call a “cataustrophe,” in some, tangles never make this move, yet the person still slides quickly into cognitive impairment, Sperling noted. Could LATE explain some of these differences?
In the HABS cohort, low hippocampal volume strongly predicted cognitive decline, even among people who had no Aβ deposition, Sperling said, hinting they might indeed have LATE, but without autopsy data or a biomarker, this is but a hunch.
Hints that LATE causes cognitive decline also came from ApoE4 carriers, who are likelier to get AD than noncarriers. While cognition in carriers declined faster than in noncarriers, in some, this happened independently of both plaques and tangles, Sperling said. Since recent studies suggest that ApoE4 boosts risk for LATE, perhaps the risk allele can speed decline via TDP-43 in people with or without AD pathology, Sperling speculated.
In toto, outlier observations such as these offer faint signals that LATE plays some kind of role in determining when cognitively normal people who test positive for amyloid—the population enrolled in secondary prevention trials—slide toward cognitive impairment.
Sans biomarkers, there is no way to directly measure TDP-43 pathology during life, hobbling researchers who want to study how LATE affects cognitive decline, or recruit participants into trials that try to prevent it.
While the biomarker search is on, researchers led by Gregory Jicha, University of Kentucky, Lexington, are relying on the process of elimination to conduct what Peter Nelson, also at U. Kentucky, described as the first-ever treatment trial for LATE (see clinical trial). It recruits people who come to the U. Kentucky ADRC with memory problems yet test negative for amyloid. Those whose total tau is elevated and hippocampal volume reduced—two markers of neurodegeneration—will be invited to enroll. The trial will test nicorandil, a vasodilator developed to treat angina and other heart conditions, as a treatment for hippocampal sclerosis of aging, a pathology that most often co-occurs with LATE (see Part 1 of this series; Nelson et al., 2014; Nelson et al., 2015; Dugan et al., 2021). The drug targets SUR2, a subunit of a potassium channel. This channel senses stress or hypoxia, and helps regulate the neurovascular unit to increase blood flow. It is encoded by ABCC9, a gene that has been identified as a risk factor for hippocampal sclerosis.
“We think that part of what turns LATE into hippocampal sclerosis is a vascular problem,” Nelson told Alzforum. In support of this idea, Nelson said that in people with LATE and hippocampal sclerosis, TDP-43 inclusions are found in glial processes around small blood vessels.
TDP-43 Biomarkers
Where does the search stand for biomarkers to help assess if TDP-43 pathology influences AD trials, and to take direct aim at it with a therapeutic? Robert Rissman of the University of California, San Diego, suggested that TDP-43 packaged inside exosomes could signal the presence of LATE in the brain. Rissman highlighted recent work indicating that misfolded TDP-43 propagates between brain cells in AD, FTD, and ALS (Oct 2018 news; Jo et al., 2020). It does so via several routes, including exosomes. Many different cell types in the brain secrete these tiny membrane packages, and some manage to find their way into the blood, where their neural origins can be deciphered with cell-type-specific markers.
Previous studies have implicated exosomes in the propagation of other proteopathic proteins, including tau and α-synuclein, though the brain origin of plasma exosomes is also dogged by some controversy (Dec 2016 news; Jun 2021 news).
Might TDP-43-laden exosomes serve as a plasma biomarker for LATE? To find out, Rissman and colleagues acquired banked plasma samples from brain donors in the University of Kentucky autopsy cohort, including 22 people with LATE and 42 without. Rissman then used an IgG1 antibody that recognizes TDP-43, regardless of its phosphorylation status, to create a custom assay. Postdoc Charisse Winston sorted exosomes in the plasma by brain-cell-type origin, cracked them open, and looked for TDP-43.
What she found was surprising, Rissman said. While neuronal exosomes contained a substantial, and similar, amount of TDP-43 regardless of whether the donor had LATE pathology, astrocytic exosomes only contained TDP-43 if they came from the blood of a person with LATE. Rissman was shocked at how well the assay differentiated people with or without the pathology, adding that only four people without LATE had any detectable TDP-43 in their astrocyte exosomes, and it was minuscule compared to the load found in people with the pathology. Levels of TDP-43 also tracked with LATE in microglial exosomes, although the differences were not as stark as those in astrocytes.
The scientists repeated the analysis with Cusabio’s commercially available test, which is based on a TDP-43 antibody that recognizes an undisclosed epitope. With this reagent company’s test, only the astrocyte TDP-43 tracked with pathology. Oddly, with the commercial kit, microglial exosomes from people without LATE had more TDP-43 than those with the pathology. Rissman has no explanation for the discrepancy, but said the homemade assay was orders of magnitude more sensitive than the commercial one.
Rissman also reported that the concentration of free TDP-43 in the plasma was similar regardless of LATE, precluding it as a biomarker for the disorder.
Nelson called the potential for a sensitive plasma biomarker for LATE “mind-blowing.” If confirmed, a plasma biomarker for TDP-43 pathology could be useful not only for LATE, but perhaps for FTD as well. TDP-43 pathology underlies about half of FTD cases, while the other half are tauopathies. A TDP-43 marker would be particularly welcome for people with sporadic FTD, who have no mutation to denote their underlying pathology. Since there is no way to tell which neuropathology lurks in their brains, plasma exosome makers could direct them toward tau- or TDP-43-specific clinical trials. Tau-PET tracers do not reliably detect the forms of tau that accumulate in FTD.
Rissman emphasized that his group's findings are preliminary, and must be repeated in larger cohorts. He plans to test plasma from people with other neurodegenerative diseases, including AD and FTD. He also wants to evaluate baseline plasma samples collected in ongoing secondary AD prevention trials, namely A4 and AHEAD 3-45, he told Alzforum. If the assay holds up, researchers will be able to determine whether the TDP-43 biomarker correlated with the trajectory of cognitive decline in placebo groups, and/or the effectiveness of amyloid-targeted therapies in treatment groups after the trials are completed.
Two other potential LATE biomarkers presented at the NIA workshop came from the ranks of structural and functional neuroimaging. Lei Wang of Ohio State University in Columbus shared preliminary data correlating the shape of the hippocampus to the presence of LATE. Using MRI scans taken of participants in the ROS-MAP cohort shortly before they died, Wang found upon autopsy that people who had had LATE tended to have an inward deformation of the hippocampus. In other words, the inner curvature of the seahorse-shaped structure was more concave. This bend correlated with the stage of LATE, and with worse scores on tests of cognitive impairment. Notably, the bend was tied specifically to TDP-43 pathology, occurring independently from hippocampal sclerosis.
Michel Grothe of the Instituto de Biomedicina de Sevilla in Spain described distinctive patterns of hypometabolism in people with LATE versus AD. Recent studies have used FDG-PET to detect metabolic changes in the brain caused by LATE or by hippocampal sclerosis (Botha et al., 2018; Buciuc et al., 2020). To investigate if FDG-PET distinguishes people with LATE and AD, Grothe analyzed scans taken before death among 30 people in the ADNI autopsy cohort, including seven with LATE and no AD pathology, and 23 with plaques and tangles but no LATE. Using these relatively “pure” cases, Grothe found a distinctive, temporolimbic pattern of hypometabolism in LATE, and a more temporoparietal pattern of metabolic dysfunction in AD. Notably, the LATE pattern was independent of hippocampal sclerosis, which was present in three cases.
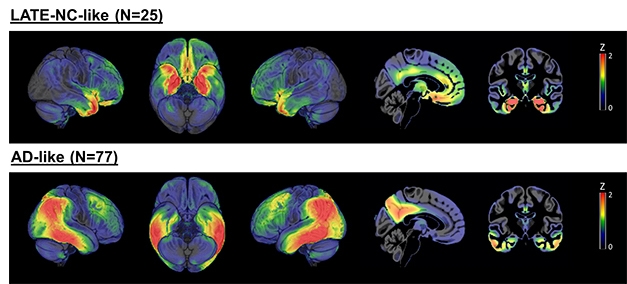
LATE versus AD. A subset of 242 people with a clinical AD dementia diagnosis had either a LATE-like pattern of hypometabolism (top) or AD-like pattern (bottom). Most of the remaining people had patterns that lay somewhere in between. [Courtesy of Michel Grothe, Instituto de Biomedicina de Sevilla, Spain.]
Going back to the whole ADNI cohort, Grothe found that among 242 participants with a clinical diagnosis of AD dementia, 25 had the LATE hypometabolism pattern, hinting that they may have been misdiagnosed, while 77 had the AD-like pattern. The remaining 140 people did not clearly fall into either category. Instead, most fell on a continuum between the AD and LATE patterns, suggesting they may have had a mix of the two pathologies, said Grothe. For the remainder, the metabolic patterns fit nowhere into the proposed continuum.
In keeping with the misdiagnosis idea, Grothe found that people who had more of a LATE pattern had declined more slowly on the MMSE, had closer to normal AD biomarkers, including CSF Aβ42 and p-tau, and were older than those who leaned toward the AD pattern. The proportion of people carrying an ApoE4 allele was smaller among LATE leaners than it was among AD leaners, and they were likelier to carry a TMEM106B risk allele, which has been tied to LATE as well as FTD.
Though he did not present it at the workshop, Grothe told Alzforum that he has examined FDG-PET patterns among people in the ADNI autopsy cohort who had died with a mix of both LATE and AD pathologies. The AD-like hypometabolism pattern predominated, as did plaques and tangles.
Though the findings don’t paint FDG-PET as a clearcut diagnostic tool for LATE, Grothe thinks the scans could prove useful, especially if the LATE to AD continuum can be more clearly defined. While acknowledging there was substantial overlap between people with LATE and AD in these structural and functional imaging measures, Nelson and other researchers were enthusiastic that they might prove valuable upon further refinement.
The meeting agenda, recording, and parallel presentations on LATE are available at LATE 2022.—Jessica Shugart
References
News Citations
- Scientists Say LATE Worsens Cognitive Decline
- Virtual Workshop Tackles LATE, a Cause of Late-life Dementia
- TDP-43 Joins Cell-To-Cell Propagation Gang
- Astrocytes and Exosomes Implicated in Protein Propagation
- Neuronal Exosomes Embroiled in Controversy
Paper Citations
- Nelson PT, Estus S, Abner EL, Parikh I, Malik M, Neltner JH, Ighodaro E, Wang WX, Wilfred BR, Wang LS, Kukull WA, Nandakumar K, Farman ML, Poon WW, Corrada MM, Kawas CH, Cribbs DH, Bennett DA, Schneider JA, Larson EB, Crane PK, Valladares O, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Scheff SW, Sonnen JA, Haines JL, Pericak-Vance MA, Mayeux R, Farrer LA, Van Eldik LJ, Horbinski C, Green RC, Gearing M, Poon LW, Kramer PL, Woltjer RL, Montine TJ, Partch AB, Rajic AJ, Richmire K, Monsell SE, Alzheimer’ Disease Genetic Consortium, Schellenberg GD, Fardo DW. ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathol. 2014 Jun;127(6):825-43. Epub 2014 Apr 27 PubMed.
- Nelson PT, Jicha GA, Wang WX, Ighodaro E, Artiushin S, Nichols CG, Fardo DW. ABCC9/SUR2 in the brain: Implications for hippocampal sclerosis of aging and a potential therapeutic target. Ageing Res Rev. 2015 Nov;24(Pt B):111-25. Epub 2015 Jul 28 PubMed.
- Dugan AJ, Nelson PT, Katsumata Y, Shade LM, Boehme KL, Teylan MA, Cykowski MD, Mukherjee S, Kauwe JS, Hohman TJ, Schneider JA, Alzheimer’s Disease Genetics Consortium, Fardo DW. Analysis of genes (TMEM106B, GRN, ABCC9, KCNMB2, and APOE) implicated in risk for LATE-NC and hippocampal sclerosis provides pathogenetic insights: a retrospective genetic association study. Acta Neuropathol Commun. 2021 Sep 15;9(1):152. PubMed.
- Jo M, Lee S, Jeon YM, Kim S, Kwon Y, Kim HJ. The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Exp Mol Med. 2020 Oct;52(10):1652-1662. Epub 2020 Oct 13 PubMed.
- Botha H, Mantyh WG, Murray ME, Knopman DS, Przybelski SA, Wiste HJ, Graff-Radford J, Josephs KA, Schwarz CG, Kremers WK, Boeve BF, Petersen RC, Machulda MM, Parisi JE, Dickson DW, Lowe V, Jack CR Jr, Jones DT. FDG-PET in tau-negative amnestic dementia resembles that of autopsy-proven hippocampal sclerosis. Brain. 2018 Apr 1;141(4):1201-1217. PubMed.
- Buciuc M, Botha H, Murray ME, Schwarz CG, Senjem ML, Jones DT, Knopman DS, Boeve BF, Petersen RC, Jack CR Jr, Petrucelli L, Parisi JE, Dickson DW, Lowe V, Whitwell JL, Josephs KA. Utility of FDG-PET in diagnosis of Alzheimer-related TDP-43 proteinopathy. Neurology. 2020 Jul 7;95(1):e23-e34. Epub 2020 Jun 9 PubMed.
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
NeuroDex INC
NeuroDex is a firm believer that diagnosis in the neurodegenerative space should move from a symptom-based diagnosis to a pathology-based diagnosis. Such a shift would increase the accuracy of treatment, clinical trials, and ensure proper detection of comorbidities. Therefore, it was with great interest that we read your commentary regarding the urgent need for TDP-43 biomarkers and the summary of the exciting LATE 2022 meeting.
NeuroDex developed a novel platform for cell-type-specific exosome isolation from plasma (including neuron- and glia-derived exosomes), which enabled us to measure TDP-43 in neuron-derived exosomes (NDEs) of patients diagnosed with ALS and FTD. As part of the project funded by a Target ALS grant, over 85 ALS patients and controls were analyzed, and TDP-43 levels were found significantly (P=0.0014) elevated in ALS patients. Furthermore, analysis of an open-label clinical trial demonstrated a correlation between the changes in NDE-associated TDP-43 and symptom progression (ALSFRS-R score slope). The mechanism of TDP-43 secretion by exosomes is not fully understood, and thus the relationship between cellular TDP-43 localization and post-translational modification and its levels in exosomes is not yet clear. However, it is reasonable to assume that cytoplasmic localization and ubiquitination increase TDP-43 secretion in exosomes.
In this light, the findings that demonstrate elevated TDP-43 in astrocyte-derived exosomes are highly encouraging. Some of our internal work, as well as studies by others, suggest an interaction between the glia and neuron-derived exosomes. For example, we found elevated levels of astrocyte-specific proteins, such as GFAP and oligodendrocyte-specific proteins, e.g. MBP, in neuron-derived exosomes, which was not the result of contamination, since proteins from other cellular sources like CD14, ICAM, albumin, and ApoA were not enriched. The underlying biology of these is the subject of further study.
An additional question is whether exosomal TDP-43 resides in the intraluminal space versus the outer membrane leaflet (surface). In the past, when using an assay that measured α-synuclein on the exosome surface, we also found a more significant elevation in microglia and oligodendrocyte-derived exosomes of Parkinson’s patients.
We hope that brain-specific exosomes (from neurons, microglia, astrocytes, oligodendrocytes) will enable blood-based stratification of dementia based on the underlying pathology. This will enable improved design of clinical trials, provide more accurate diagnosis and prognosis for patients, and avoid prescription of unhelpful or even harmful treatments. The recent exciting finding of TDP-43 in astrocyte-derived exosomes as a biomarker for LATE represents the first steps in this direction, and we are looking forward to the continued development of this biomarker platform.
Make a Comment
To make a comment you must login or register.