Astrocytes and Exosomes Implicated in Protein Propagation
Quick Links
Many researchers now believe that misfolded proteins spread from cell to cell across the brain, corrupting normal proteins as they go, yet exactly how this propagation would happen remains unclear. At the Society for Neuroscience annual meeting, held November 12-16 in San Diego, scientists identified specific mechanisms that may be involved in the spread of pathological proteins in Parkinson’s and Alzheimer’s diseases. They argued that astrocytes transmit α-synuclein aggregates by cell-to-cell contact; for tau, they argued that its trans-synaptic spread depends on the secretory vesicles known as exosomes. In addition, researchers debuted a new method for following tau aggregates over time in living mouse brain.
“Up until a few years ago, we did not understand how Parkinson’s disease progresses at the molecular level,” noted Alice Chen-Plotkin of the University of Pennsylvania, who moderated an SfN press conference on Parkinson’s research. “Now, we may be starting to discover how the disease worsens, which could give insights into ways to slow it down or stop it,” she wrote to Alzforum.
Numerous studies have found that misfolded proteins can seed aggregation of normal protein throughout animal brains, and that such seeds may even be transferred from one person to the next in tissue transplants (see Apr 2015 conference news; Jan 2016 news). Many proteins fold into specific shapes, or “strains,” which propagate in vitro and give rise to distinct pathologies and symptoms in animal models (see Sep 2013 news; Jun 2015 news; Nov 2016 news). However, some researchers have pointed out that existing data fall short of demonstrating a causal role for protein propagation in human disease, or showing how proteins travel through tissue (see Apr 2016 webinar).

Glial Indigestion.
Cultured astrocyte (green) has ingested recombinant α-synuclein (red), which then accumulates around its nucleus (blue). [Courtesy of Jinar Rostami and Anna Erlandsson.]
Researchers led by Anna Erlandsson of Uppsala University, Sweden, working with Laurent Roybon of Lund University, also in Sweden, wondered whether astrocytes might aid and abet the migration of pathological proteins. Some recent studies have found that these glial cells pass misfolded prion protein to neurons (see Hollister et al., 2015; Victoria et al., 2016). At an SfN press conference, Jinar Rostami in Erlandsson’s group made the case that astrocytes transmit α-synuclein aggregates as well.
The researchers tested for protein spread using astrocytes derived from human pluripotent stem cells. Some researchers have pointed out that cultured astrocytes resemble immature or reactive cells more than mature brain astrocytes. However, the iPSC-derived astrocytes expressed markers for mature astrocytes, such as S100β, but low levels of proteins that mark reactive astrocytes, such as GFAP, Rostami told Alzforum. Rostami added recombinant monomeric or oligomeric α-synuclein to these cultures. Astrocytes gobbled up the proteins. They quickly degraded the monomers, but oligomeric α-synuclein was a different story. It lingered inside cells, passing through lysosomes to end up in the trans-Golgi network around the nucleus (see image above). After six days, astrocytes containing α-synuclein aggregates betrayed signs of cellular stress, including fragmented mitochondria and swollen endoplasmic reticulum. The cells began to pass the α-synuclein aggregates off to other astrocytes, as though playing a cell-culture game of “hot potato.” Time-lapse microscopy revealed instances of astrocytes making direct contact with neighboring cells to transfer labeled aggregates. In other cases, cells formed long, thin tunneling nanotubes to reach more distant cells (see Gerdes et al., 2013). Aggregates traveled down these tubes. Adding an inhibitor of cytoskeletal reorganization to the cultures to suppress contacts between cells cut α-synuclein transfer in half, Rostami reported in San Diego.
Does this happen in the Parkinson’s disease brain? This is unknown, but Rostami plans to look in vivo in mouse models. Meanwhile, she believes α-synuclein released by dying neurons in the midbrain of Parkinson’s patients could be taken up by astrocytes there and spread further. Erlandsson noted that the data may have therapeutic implications. “If we could stimulate degradation of α-synuclein aggregates by astrocytes, it could reduce spreading,” she told Alzforum. Other researchers consider this a challenging proposition since at least in neurons, α-synuclein resists autophagy (see Apr 2013 news).
That said, one potential treatment being tested in Parkinson’s trials, the cancer drug nilotinib, enhances autophagic removal of the protein by blocking Abl, a kinase that protects synuclein from degradation (see Nov 2015 conference news).
Speaking at the same press conference, Collin Challis, working with Viviana Gradinaru of the California Institute of Technology, Pasadena, reported that α-synuclein deposits added to the intestines of rats stimulate misfolding of endogenous α-synuclein there. Endogenous, but now misfolded, synuclein then migrates up the vagal nerve to the brainstem and midbrain, where it seems to affect behavior. Challis’ findings dovetail with the hypothesis that α-synuclein aggregation can start in the gut in Parkinson’s disease, and from there propagate to the brain (see Jul 2011 news series; Oct 2016 news; Holmqvist et al., 2014).
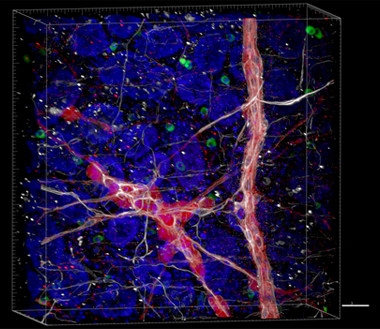
From Gut to Brain?
Tissue clearing reveals rare α-synuclein fibrils (green) in the endothelial cells (nuclei shown in blue) of the gut lining, and their three-dimensional relationship with neurons (red) of myenteric plexus ganglia and astrocytes (white). [Courtesy of Collin Challis and the Gradinaru Group at Caltech.]
Challis and colleagues injected preformed fibrils of recombinant α-synuclein into the stomach and gut lining of 22 adult wild-type mice, with another 19 receiving control saline/BSA injections. Within one week, the former developed gastrointestinal problems, defecating more than control mice. This correlated with the accumulation of endogenous α-synuclein in the gut lining, as seen by staining with an antibody that detects α-synuclein phosphorylated at serine 129, but not the recombinant preformed fibrils, which remain unphosphorylated (see image above). Phosphorylation at S129 promotes oligomerization and probably does not occur after fibrils form, Gradinaru told Alzforum (see Volpicelli-Daley et al., 2011; Samuel et al., 2016). Fibril formation in the gut peaked at three weeks, then returned to normal by two months, and gastrointestinal dysfunction followed the same time course. Meanwhile, endogenous α-synuclein aggregates appeared in the brainstem by three weeks, and the midbrain by two months. At this later time point, the mice began having trouble turning around on a vertical pole, indicating muscle weakness or loss of motor control. The results support the idea that the propagation of misfolded α-synuclein from gut to brain causes disease symptoms, Challis said. He did not address whether astrocytes in the brain or the enteric plexus played any role in the spread of α-synuclein.
Once protein aggregates have reached neurons, how might they travel among them? Some recent research suggests that exosomes smuggle proteins out of one and into another (see Dec 2014 conference news; Oct 2015 news). In San Diego, Yipeng Wang of the German Center for Neurodegenerative Diseases (DZNE), Bonn, presented evidence supporting the idea that tau migrates in this fashion. Working with Eckhard and Eva-Maria Mandelkow at DZNE, Wang first characterized exosomes secreted by primary cortical neurons cultured from wild-type rats. He found that these exosomes contained full-length tau that remained largely unphosphorylated. Tau likely rode inside the vesicles, rather than on their surface, because proteinase K failed to digest the protein when added to intact exosomes.
The researchers then examined whether these exosomes could transfer tau between cells. Wang collected exosomes secreted by a mouse neuroblastoma cell line that expressed tau labeled with GFP, and added the exosomes to two-chambered microfluidic culture devices containing mouse hippocampal neurons. Each such neuron extended an axon through a microgroove to contact the neuron in the adjoining chamber. When Wang added the exosomes after four days of microfluidic culture, before the neurons had formed synapses, no tau passed to their neighboring cells. After 11 days in culture, however, once synapses had formed, human tau appeared in the distant neuron, indicating that functional synapses are required to transfer tau. Naked tau from sonicated exosomes added directly to the chamber did not transmit to the neighboring cell, suggesting that intact exosomes are required. Furthermore, Wang reported that depolarization of the cultured neurons stimulated exosome release, suggesting these vesicles are shed in some sort of regulated physiological process. Wang did not speculate on why that might be.
Does this exosomal transport play a role in transmitting aggregates of tau? Wang and colleagues expressed an aggregating form of tau, DK280, in the mouse neuroblastoma cells, and found that the aggregates passed among them, again via exosomes. This experiment did not use hippocampal neurons in the microfluidic chamber.
Could neurons from people do the same? To begin to test this, the researchers isolated exosomes from the cerebrospinal fluid of Alzheimer’s patients and from healthy controls. The exosomes from patients contained tau oligomers. When added to the mouse neuroblastoma cells that expressed DK280 tau, the human exosomes promoted tau aggregation. Exosomes mediate the spreading of tau, Wang concluded.
Aggregated Aβ, as well, may travel in exosomes. Working with Martin Hallbeck of Linköping University, Sweden, Maitrayee Sinha isolated exosomes from postmortem Alzheimer’s brain tissue. Sinha reported in San Diego that these exosomes contained more oligomeric Aβ, as judged by binding to conformation-specific antibodies, than those from age-matched control tissue. When she added these exosomes to human neuronal cultures generated from induced pluripotent stem cells, the Aβ oligomers were taken up and passed to neighboring cells. Neurons containing the aggregates began to die. The researchers were able to prevent Aβ transfer either by inhibiting endocytosis or by silencing genes involved in exosome production and secretion, demonstrating the key role of exosomes in this process.
A Window on Live Aggregation
Because following protein aggregation and propagation in vivo is technically challenging, most of the data in this line of research come from in-vitro studies. That said, Sarah DeVos and Brad Hyman of Massachusetts General Hospital, Charlestown, have devised a way to follow tau aggregates in the living brain. At SfN, DeVos described how she modified a fluorescence resonance energy transfer (FRET) assay developed by Marc Diamond at the University of Texas Southwestern Medical Center, Dallas (see Sep 2013 news). She incorporated a mutant human tau repeat domain labeled with cyan fluorescent protein along with another one labeled with yellow fluorescent protein in the same viral vector. DeVos tested the vector in primary neurons, and found that as predicted, transfected neurons expressed equal amounts of both types of labeled tau repeat. When the repeats aggregated, they produced the FRET signal.
The researchers then injected the vector into layer II-III neurons in the cortices of adult Tg4510 mice, which express human mutant P301L tau. Imaging the neurons through a cranial window, they saw the formation of tau aggregates in cells that took up the vector (for a three-dimensional visual of this, see the video clip below). With this system, the researchers can now track the same neuron over months, DeVos told the audience. She wants to study the fate of transfected neurons, for example by analyzing their gene expression to see how the presence of tau aggregates alters them. An audience member pointed out that the FRET assay merely detects aggregates, not necessarily true fibrils. DeVos said she will use electron microscopy to confirm the nature of the aggregates. She also intends to track whether aggregates are transported down axons, although the spatial limitations of the cranial window will prevent directly seeing transfer to distant projection areas.—Madolyn Bowman Rogers
Media
References
News Citations
- Protein Propagation Real, but Mechanisms Hazy
- News Brief: More Evidence for Aβ Spread Between People
- Does Aβ Come In Strains? Glimpse Into Human Brain Suggests Yes
- Shape of α-Synuclein Aggregates Influences Pathology
- More Evidence That Distinct Tau Strains May Cause Different Tauopathies
- Synuclein Resists and Disrupts Autophagy
- Potential Parkinson’s Treatments Target α-Synuclein, Cell Replacement
- Parkinson’s: Thinking Outside the Brain’s Black Box
- Could Bacterial Amyloid Trigger Parkinson’s Pathology?
- Exosomes: Purveyors of Neurodegenerative Disease?
- Deadly Delivery: Microglia May Traffic Tau Via Exosomes
- Antibodies Stop Toxic Tau in Its Extracellular Tracks
Webinar Citations
Research Models Citations
Paper Citations
- Hollister JR, Lee KS, Dorward DW, Baron GS. Efficient uptake and dissemination of scrapie prion protein by astrocytes and fibroblasts from adult hamster brain. PLoS One. 2015;10(1):e0115351. Epub 2015 Jan 30 PubMed.
- Victoria GS, Arkhipenko A, Zhu S, Syan S, Zurzolo C. Astrocyte-to-neuron intercellular prion transfer is mediated by cell-cell contact. Sci Rep. 2016 Feb 9;6:20762. PubMed.
- Gerdes HH, Rustom A, Wang X. Tunneling nanotubes, an emerging intercellular communication route in development. Mech Dev. 2013 Jun-Aug;130(6-8):381-7. Epub 2012 Dec 14 PubMed.
- Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Björklund T, Wang ZY, Roybon L, Melki R, Li JY. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014 Dec;128(6):805-20. Epub 2014 Oct 9 PubMed.
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011 Oct 6;72(1):57-71. PubMed.
- Samuel F, Flavin WP, Iqbal S, Pacelli C, Sri Renganathan SD, Trudeau LE, Campbell EM, Fraser PE, Tandon A. Effects of Serine 129 Phosphorylation on α-Synuclein Aggregation, Membrane Association, and Internalization. J Biol Chem. 2016 Feb 26;291(9):4374-85. Epub 2015 Dec 30 PubMed.
External Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.