Mouse Tau Knock-Ins: A Tale of Two Pathologies
Quick Links
Creating models for primary tauopathies sounds simple enough. Select a human tau gene variant that causes disease, stick it where the endogenous mouse gene sits in its genome, then wait a few months for tangles to gum up the brain. In practice, this has been more a nightmare than a dream. As scientists outlined at the Tau Global Conference, held April 24-25 in London, pathology in tau knock-in mice is often weak or non-existent. When it does appear, it can be extremely slow, crop up in unexpected places, and differ from the molecular changes to tau seen in people. In all these respects, knock-ins can look dramatically different from each other.
- Scientists have replaced mouse tau with various human tau variants.
- These knock-ins don’t fully recapitulate human tauopathies.
- Some are “phosphorylaters,” others are “seeders.”
Can the field make a good mouse? “We are not there yet,” Karen Duff, University College London, told a packed room at the Hilton Park Lane, after she outlined the differences between tau knock-ins. “We don’t understand the events going on in these mice.”
To be sure, that human variants evoke different phenotypes in mice comes as no surprise. Primary tauopathies themselves are notoriously heterogeneous, with different fibril structures that accumulate in different parts of the brain, causing different symptoms. “We know the heterogeneity translates to the clinic, which is why we can’t do basket trials in primary tauopathies,” Kristin Wildsmith, Eisai Inc. Nutley, New Jersey, told Alzforum. Basket trials test one drug in multiple diseases. Platform trials test multiple drugs in the same disease; one is slated to begin this year for tauopathies (see Part 2 of this series). “A key message is that the genetic mutations have different effects and yield heterogenous tau pathologies. This is different from mutations that cause dominantly inherited Alzheimer’s disease,” Wildsmith said.
Despite these challenges, the sense in London was that scientists are starting to figure out how different MAPT variants conjure different pathologies. Specific mutations, post-translational modifications, proteolytic processing, and/or structural changes all play a part. “With new generations of models, we hope to tease this all out,” said Duff.
All Models Aren’t Created Equal
For years, scientists relied on MAPT overexpression to study tauopathy. The caveats are known: disruption of endogenous mouse genes, clobbering mouse neurons with up to 70 copies of human tau, and making the protein in the wrong place, i.e., the soma rather than the axon. In 2019, enter knock-ins. Scientists led by Takaomi Saido, RIKEN, Wako-City, Japan, introduced the human wild-type gene into the mouse MAPT locus (Saito et al., 2019; Hashimoto et al. 2019).
Late last year, scientists led by Duff and Saido debuted two new knock-ins. One carries a G to A mutation in intron 10 of the MAPT gene, and the other combines this Int10+3 G>A variant with the S305N mutation. Both these mutations cause frontotemporal dementia.
More recently, these collaborators, led by Saido and Naoto Watamura, also at RIKEN, introduced triple transgenic mice that add the FTD variant S320F (May 2025 news). Meanwhile, Duff and colleagues have been characterizing knock-ins carrying the Int10+3 G>A variant with the P301S mutation.
If that’s not enough, scientists in the U.S. led by Michael Koob, University of Minnesota, Minneapolis, have generated seven gene replacement (GR) mouse lines that have their endogenous tau swapped with a section of human chromosome 17 containing wild-type MAPT or one of give genetic variants (Benzow et al., 2024)—P301L, N279K, L266V, G272V, and a C to T point mutation in the intron following exon 10 (IVS10+16T).
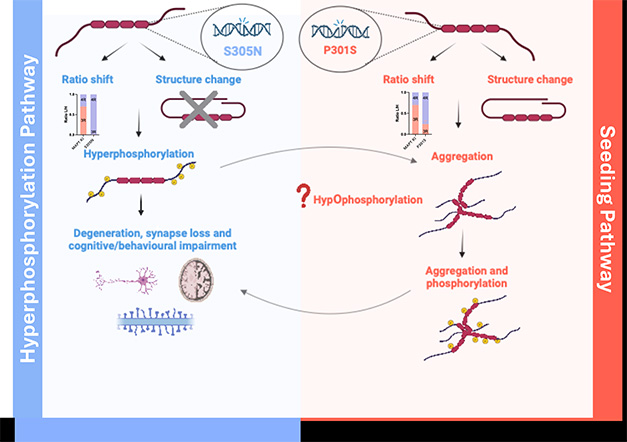
Two Tau Paths. In knock-in mice, tau pathology follows either of two paths. Some mice hyperphosphorylate the protein (left), others form aggregates that seed fibrils (right). [Created in BioRender. Foiani, M. (2025.)]
Phosphorylation Does Not Equal Aggregation
What happens in the brains of all these mice? In London, Duff reported that they seem to fall broadly into one of two categories. They start out being either hyperphosphorylaters or seeders. The former include S305N, possibly GR-N279K, and GR-10+16 variants. Using mass spectrometry, Raja Nirujogi at the University of Dundee, Scotland, and Martha Foiani in Duff’s lab measured phosphorylation at dozens of tau sites in S305N mice, yet found no tau in the brain that was able to seed aggregation in cell assays. Even if they aged mice to 30 months: no seeds. Still, this mutation did correlate with loss of synapses.
Foiani is also studying the GR mice from Koob’s lab, which are yet to be described in the literature but are available from Jackson Laboratory, Bar Harbor, Maine. Early data suggests that 10+16 mice also hyperphosphorylate tau 202, as do N279K mice, albeit less robustly. Neither mouse seems to generate tau species that can seed aggregates—at least by 12 months of age.
RIKEN’s P301S knock-ins, on the other hand, barely phosphorylated tau. Mass spec showed that on many threonines and serines, wild-type mice did a much better job of this than did the knock-ins. Same story in immunoblots, where antibodies detected almost no phosphorylation at S202, T205, S396, or S404—sites where tau is typically hyperphosphorylated. Where the P301S did shine, however, was in producing seeds. These were detectable in the brain as early as six months of age, said Duff. Late in life, around 25 months, these mice formed tangles, but still had no neurodegeneration.
Based on the data thus far, Duff sees two pathways to early tau pathology typified by the S305N and P301S mutations. The former hyperphosphorylates tau but seeds no aggregates, while the latter hypophosphorylates tau initially, seeds aggregates, and later makes fibrils. Duff suspects these pathways converge as the mice age, whereby the hyperphosphorylated tau eventually forms aggregates and tangles, whereas the seed-competent models eventually become hyperphosphorylated and cause neurodegeneration.
The presentation sparked lively discussion. Wildsmith was fascinated that mutations so close together in the protein, at positions 301 and 305, could evoke such different sequences of events. Nathan Smith, Rensselaer Polytechnic Institute, New York, thought it strange that phosphorylation and aggregation seemed to be distinct. “Have you looked at any other modifications in the 301S mutants that explain seeding?” he asked. Duff has not, but wants to study the full suite of post-translational modifications.
To one attendee, the data challenges current dogma on fibrillization. “We know that phosphorylation produces the core fold we see in cryoEM, but what you are saying seems to run counter to that,” she said. Duff agreed, but noted the cryoEM structures came from tau that had been phosphorylated in vitro (Chakraborty et al., 2024). Duff sees phosphorylation and seeding in vivo, so she does not consider the comparison fair.
Koob, who missed the London meeting, explained his GR mice to Alzforum. “We are trying to model the genetics of dementia,” he said. “So we have replaced the whole mouse tau region with the whole human tau region.” He said the Saido lab swapped in a smaller region, i.e., exons 1 through 14, hence the two sets of mice don’t directly compare. Nevertheless, Koob confirmed largely what the other groups are seeing, namely that knocking in a single pathogenic variant evokes little pathology. “The expectation was that tau mutations would lead to early tau proteinopathy, but that is not what we are finding,” he said.
Koob does see some phospho-tau, in the right neurons, if mice live to 28 months, but not consistently so. “From three siblings at that age, one had more progressive pathology. This is not how we want models to work in practice,” said Koob. He thinks time is an important element. “We don’t see tau pathology in children, and since mice only live two to three years, maybe there’s just not enough time for the pathology to emerge,” he said. When asked what her ideal model would be, Duff said, “A mouse that lives beyond 2½ years.”
Koob says aging is why it makes some sense to piggyback variants in a single model, as Saido and colleagues have done. He sees those as pathology rather than genetic models. His solution? An environmental trigger, such as a blow to the head. The idea with this injury is to stretch the axon. “Then we can see tau pathology in really young mice,” with thousands of neurons containing tangles after just one week, he told Alzforum. He thinks human neurons accumulate tau pathology partly because they have longer and more fragile axons.
That makes some sense to Kim Green and Grant MacGregor at University of California, Irvine. “It seems that mice are more resilient than humans,” MacGregor told Alzforum. There could be many reasons why. MacGregor thinks the mice’s higher metabolic rate, higher capacity to endure damage, or even a different lipid composition might explain it. Green sees parallels with models of sporadic AD. “If you humanize mouse amyloid, the animals will not develop pathology. You have to push the system with mutations to get them to produce plaques,” Green said.
Like Koob, MacGregor and Green also made tau knock-in models as part of the MODEL-AD project to study late-onset AD (Sep 2024 conference news). They took a similar approach, swapping in not only the human coding region but also the upstream paraphernalia that helps regulate gene expression, including promoters and genes for natural antisense transcripts (Simone et al., 2021). They used wild-type and FTD variants. These tau knock-ins are still too young to check for pathology.
For her part, Judith Steen, Children’s Hospital Boston, thinks mice only partially recapitulate human tauopathies because they modify the protein differently. She uses mass spectrometry to track proteomics and post-translational modifications that might be involved in pathogenesis. Unsurprisingly, Steen finds that human tau looks different than tau in mouse overexpression models. Although the order of phosphorylation is similar, ubiquitination and acetylation are not. In people, both occur in late-stage disease, whereas in mice they never do (Wenger et al., 2023). Even between people with AD, post-translational modification differ, and may explain why some decline more rapidly (Jun 2020 news). Steen believes that getting mice to modify tau the right way could lead to better models.
Similarly, she asks if the tau proteome might help explain heterogeneity across the different tauopathies. She studies this question using brain tissue, CSF, and plasma. Among a cohort of more than 200 volunteers, Steen sees more tau oligomers in PSP than in other tauopathies. In other words, in PSP there is more toxicity with less aggregation. This may help explain why scientists have found it hard to detect tau aggregates in PSP using PET ligands that work for AD (Jul 2024 news). “A PET agent for PSP will have to bind 10- to 60-fold tighter than do those for AD tangles,” said Steen.
While PSP tau may be the most potent at forming tangles, Steen found that AD and chronic traumatic encephalopathy come with the most post-translational modifications of tau. To define differences between tauopathies in more detail, she plans to apply single-cell and spatial proteomics with the most sensitive instruments. “We can now analyze up to 3,000 proteins per cell at one time,” she said. Steen hopes this will help identify how tau variants alter cell biology, which, in turn, could suggest new ways to model disease and help design better treatments. “Drug design depends on knowing how much bad tau you have to tackle,” she said.—Tom Fagan
References
News Citations
- Next Up—Combination Trials for Sporadic Alzheimer’s
- Triple Trouble: New Knock-in Turbocharges Tauopathy
- MODEL-AD Probably Has Something for You
- Tau: Why Alzheimer’s Worsens Fast in Some, Slowly in Others
- PET Tracers For Non-Alzheimer’s Tauopathies Enter Clinical Testing
Mutations Citations
- MAPT IVS10+3 G>A
- MAPT S305N
- MAPT S320F
- MAPT P301S
- MAPT P301L
- MAPT N279K
- MAPT L266V
- MAPT G272V
- MAPT IVS10+16 C>T
Paper Citations
- Saito T, Mihira N, Matsuba Y, Sasaguri H, Hashimoto S, Narasimhan S, Zhang B, Murayama S, Higuchi M, Lee VM, Trojanowski JQ, Saido TC. Humanization of the entire murine Mapt gene provides a murine model of pathological human tau propagation. J Biol Chem. 2019 Aug 23;294(34):12754-12765. Epub 2019 Jul 4 PubMed.
- Hashimoto S, Matsuba Y, Kamano N, Mihira N, Sahara N, Takano J, Muramatsu SI, Saido TC, Saito T. Tau binding protein CAPON induces tau aggregation and neurodegeneration. Nat Commun. 2019 Jun 3;10(1):2394. PubMed.
- Benzow K, Karanjeet K, Oblak AL, Carter GW, Sasner M, Koob MD. Gene replacement-Alzheimer's disease (GR-AD): Modeling the genetics of human dementias in mice. Alzheimers Dement. 2024 Apr;20(4):3080-3087. Epub 2024 Feb 11 PubMed.
- Chakraborty P, Ibáñez de Opakua A, Purslow JA, Fromm SA, Chatterjee D, Zachrdla M, Zhuang S, Puri S, Wolozin B, Zweckstetter M. GSK3β phosphorylation catalyzes the aggregation of tau into Alzheimer's disease-like filaments. Proc Natl Acad Sci U S A. 2024 Dec 24;121(52):e2414176121. Epub 2024 Dec 18 PubMed.
- Simone R, Javad F, Emmett W, Wilkins OG, Almeida FL, Barahona-Torres N, Zareba-Paslawska J, Ehteramyan M, Zuccotti P, Modelska A, Siva K, Virdi GS, Mitchell JS, Harley J, Kay VA, Hondhamuni G, Trabzuni D, Ryten M, Wray S, Preza E, Kia DA, Pittman A, Ferrari R, Manzoni C, Lees A, Hardy JA, Denti MA, Quattrone A, Patani R, Svenningsson P, Warner TT, Plagnol V, Ule J, de Silva R. MIR-NATs repress MAPT translation and aid proteostasis in neurodegeneration. Nature. 2021 Jun;594(7861):117-123. Epub 2021 May 19 PubMed.
- Wenger K, Viode A, Schlaffner CN, van Zalm P, Cheng L, Dellovade T, Langlois X, Bannon A, Chang R, Connors TR, Oakley D, Renard B, Rappsilber J, Hyman B, Steen H, Steen JA. Common mouse models of tauopathy reflect early but not late human disease. Mol Neurodegener. 2023 Feb 2;18(1):10. PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.