New Evidence Confirms TREM2 Binds Aβ, Drives Protective Response
Quick Links
Variants in the microglial receptor TREM2 heighten the risk for neurodegeneration, though exactly how they do that has remained unclear as initial research produced conflicting results. Now, however, researchers appear to be reaching a consensus that TREM2 protects the brain, and that the disease-causing variants all disrupt its function in some fashion. This weakens microglia through multiple pathways, including survival, migration, and phagocytosis.
“TREM2 acts as a signaling hub,” Christian Haass of the German Center for Neurodegenerative Diseases (DZNE), Munich, explained to Alzforum. At the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29 to April 2 in Vienna, researchers added more spokes to the paradigm. Speakers agreed that TREM2 activation triggers microglia to clean up messes in response to damage or disease. Some speakers said that both oligomeric Aβ and ApoE play a role in switching on this response, while others detailed how TREM2 variants linked to Alzheimer’s disease disturb it. However, it remains unclear at what point in disease TREM2 activity does the most good, and how it could be manipulated therapeutically.
“Great strides have been made in our understanding of TREM2 in the context of AD in just a short time since the initial description of AD-associated TREM2 variants,” wrote David Holtzman and colleagues at Washington University in St. Louis in an April 19 Neuron review article. So far, most of the evidence points toward TREM2 orchestrating the microglial response to amyloid pathology, they added.
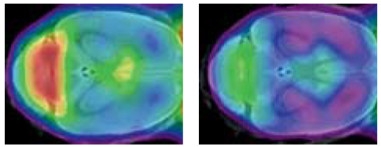
Idling Microglia.
In year-old wild-type mice (left), microglia rev up their activity (red), but in TREM2 mutant knock-ins (right), the cells remain stuck in neutral. [Courtesy of Matthias Brendel and Christian Haass.]
Interest in TREM2 took off when geneticists pinned a tripled risk of AD on the R47H variant and linked other missense mutations such as T66M with frontotemporal dementia (see Oct 2012 news; Nov 2012 news). Researchers speculated that the receptor might aid phagocytosis, but initial studies disagreed on this, with some reporting no change in plaque load in TREM2 knockouts, while others did see changes in amyloid after TREM2 stimulation (see Jun 2014 news; Jul 2016 news; Dec 2016 conference news). Since then, scientists have developed a more nuanced view, agreeing that the effects of TREM2 depend on the stage of the disease. In several studies, AD mouse models with impaired TREM2 function accumulate fewer plaques than controls at four months of age, but a heavier load by eight months (see Jul 2016 conference news).
In Vienna, Peter St. George-Hyslop of the University of Toronto dug further into the relationship between plaques and phagocytosis. He wondered if instead of interacting with plaques themselves, TREM2 might sense something that leaches off plaques, such as Aβ oligomers. Supporting this idea, he found that synthetic Aβ co-immunoprecipitated with TREM2, an interaction that could be displaced by the presence of TREM2-blocking antibodies. The bound Aβ was oligomeric, as determined by two-color coincidence detection, a method that discriminates between monomers and oligomers. Binding of these oligomers to TREM2 triggered cleavage of the receptor in a dose-dependent fashion. This receptor cleavage released a soluble N-terminal fragment, sTREM2, whereas the C-terminal fragment was internalized. TREM2 processing seemed to stimulate phagocytosis, as incubating microglia with Aβ oligomers primed them to later scarf up debris from dead neurons. Microglia lacking TREM2 did not dial up phagocytosis. The results suggest TREM2 is needed to turn on phagocytosis in response to aggregated, toxic Aβ, St. George-Hyslop said.
Previous studies had found that TREM2 bound to Aβ complexed with LDL, but had not reported an interaction with Aβ alone (see Jun 2016 conference news). It is unclear if those studies examined oligomeric Aβ, however. In addition, prior studies focused on microglia engulfing Aβ through a TREM2-mediated process, but had not shown a role for Aβ itself in activating phagocytosis, Haass noted.
How would the AD variant R47H affect this process? Examining microglia from R47H knock-in mice, St. George-Hyslop found that they gobbled up fluorescent beads efficiently and signaled normally through TREM2 and its co-receptor DAP12. However, the R47H mutants only weakly bound Aβ oligomers. As a consequence, they released less sTREM2 and phosphorylated less DAP12 in response to the oligomers. Thus, the mutant microglia poorly activate phagocytosis in the presence of Aβ, St. George-Hyslop said. The results suggest that R47H is a hypomorph, meaning it causes a partial loss of function. This distinguishes it from variants that cause FTD, which never reach the cell surface and act as complete loss-of-function mutations (see Jul 2014 webinar; Apr 2015 conference news). The findings fit with previous work that found weak binding of the R47H variant, as well as other AD-causing variants such as R62H, to ApoE and cell-surface proteoglycans, too (see Kober et al., 2016).
The H157Y mutation, prevalent in Han Chinese, drives up AD risk 11-fold (see Jiang et al., 2016). In contrast to R47H, which attenuates sTREM2 shedding, this mutation heightens it, St. George-Hyslop said. Normally, the metalloprotease ADAM10 clips membrane-bound TREM2 right after histidine 157, the site of this mutation, with cleavage typically occurring within one hour. St. George-Hyslop found that microglia expressing H157Y shed sTREM2 sooner than this, and that the soluble and C-terminal fragments of the protein build up. Moreover, this cleavage was not inhibited by the ADAM10 inhibitor batimastat, suggesting a new metalloprotease might be responsible, although cleavage occurs at the same site. Haass said he has similar experimental results. His group also sees a rise in sTREM2 in H157Y microglia at the expense of cell-surface TREM2. The end result is a loss of microglial TREM2 signaling, just as with the R47H mutation, rendering H157Y effectively a hypomorph as well, Haass said.
“The ability of TREM2 variants to enhance or impair TREM2 signaling suggests that altered TREM2 homeostasis has serious consequences in regard to the development of AD,” Holtzman and colleagues wrote in their review.
While these hypomorphs provide clues to AD mechanisms, the complete loss-of-function variants draw a clearer picture of what TREM2 does. Haass shared data from studies in a T66M knock-in mouse, which expresses no cell-surface TREM2 receptor (see Sep 2016 conference news). He found the microglia to be incapable of activating properly, and prone to dying. Whereas wild-type mice continuously dial up microglial activation with age, the T66M microglia experienced but a blip at eight months, falling back to baseline by one year of age (see image above). These passive microglia were unable to perform many normal jobs. They put out fewer processes than their wild-type counterparts. The T66M knock-in mice also expressed fewer chemotactic proteins in the brain, which attract other microglia to damaged tissue. As a result, microglia neither moved toward apoptotic neurons in aging T66M brains, nor cleaned up damaged myelin. When these knock-ins were crossed with AD mouse models, the offspring had fewer microglia clustering around plaques. In essence, knocking out TREM2 locks microglia into a homeostatic torpor, such that they can no longer respond to external stimuli and activate normally to protect the brain, Haass concluded. The data are in press at EMBO Reports.
Oleg Butovsky at Brigham and Women’s Hospital, Boston, also believes that TREM2 helps rouse microglia out of homeostasis. When microglia chew up apoptotic cells, exposed phosphatidylserines in the damaged membranes bind and activate TREM2, he said. TREM2 signaling switches on a distinct pattern of gene expression, with ApoE being the most upregulated protein. Butovsky labeled this gene expression signature MGnD. It associates with disease, with MGnD microglia being the ones that cluster around neuritic plaques.
ApoE itself appears to counter the homeostatic phenotype, Butovsky found. Inducing ApoE jolted microglia out of homeostasis, while deleting the gene returned them to a quiescent state. Butovsky did not discuss whether the E4 allele affects this process differentially, though other researchers at AD/PD reported that ApoE4 sends microglia into overdrive in response to tau (see Apr 2017 conference news). Butovsky and colleagues were also able to return activated microglia to homeostasis in AD mice by ablating TREM2. As other groups have found, the lack of TREM2 led to fewer plaques at younger ages and to more plaque at late stages. On the other hand, in P301S tauopathy mice, ablating microglial ApoE lessened damage, implying that keeping microglia in a homeostatic state helped in this condition. Other work suggests that activated microglia exacerbate tau pathology (see Maphis et al., 2015). Whether or not microglial homeostasis is beneficial may depend on the stage and type of disease, Butovsky suggested.
Karel Otero of Biogen Idec, Boston, focused instead on TREM2’s role in promoting the survival of microglia. He noted that in healthy aging, fewer than 10 percent of microglia become dystrophic, whereas in AD patients, more than half degenerate. Likewise, TREM2 knockout mice display massive numbers of dying microglia. Loss of TREM2 also impairs microglial activation, proliferation, and migration in several other models of brain damage, such as stroke, toxin-induced demyelination, and prion infection.
How does TREM2 facilitate survival? Previous work suggested that this receptor cooperated with growth factor receptor colony stimulating factor 1 (CSF-1R), also located on microglia, to enhance its signaling (see Feb 2015 conference news). Otero therefore investigated whether TREM2 might act as a co-receptor for CSF-1R ligands. Although TREM2 did not bind to CSF-1R ligands by itself, TREM2-blocking antibodies did suppress CSF-1R signaling. In addition, once CSF-1R signaling was active, TREM2 and CSF-1R co-immunoprecipitated. The data suggest that TREM2 forms a complex with activated CSF-1R and amplifies the effects of its signaling to keep microglia alive, Otero said. Several groups have reported that CSF-1R’s ligands, CSF-1 and IL34, protect against amyloid pathology in mice (e.g., Boissonneault et al., 2009; Luo et al., 2013).
In Vienna, Otero reported that disease-causing TREM2 variants disrupt this process. For example, R47H does not bind CSF-1R. In mice carrying this variant, Otero found fewer dividing and more apoptotic microglia, suggesting harm to both proliferation and survival. Crossing Tg2576 mice with TREM2 knockouts, Otero found fewer microglia around plaques and worse performance in fear-conditioning tests in the offspring. His data, too, support a model where TREM2 is neuroprotective, and its loss leads to microglial dysfunction and neurodegenerative disease, Otero concluded.—Madolyn Bowman Rogers
References
News Citations
- Mutations in TREM2 Cause Frontotemporal Dementia
- Enter the New Alzheimer’s Gene: TREM2 Variant Triples Risk
- TREM2 Mystery: Altered Microglia, No Effect on Plaques
- TREM2 Helps Phagocytes Gobble Up Aβ Coated in Antibodies
- Inflammation Helps Microglia Clear Amyloid from AD Brains
- Unbiased Screen Fingers TREM2 Ligands That Promote Aβ Uptake
- Keystone Meeting on Microglia/Neurodegeneration: Here’s the Buzz
- Microglia—Who Are You Really? New Clues Emerge
- New Data Reinforces Concept of Protein Propagation
- ApoE and Tau: Unholy Alliance Spawns Neurodegeneration
- United in Confusion: TREM2 Puzzles Researchers in Taos
Webinar Citations
Research Models Citations
Paper Citations
- Kober DL, Alexander-Brett JM, Karch CM, Cruchaga C, Colonna M, Holtzman MJ, Brett TJ. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife. 2016 Dec 20;5 PubMed.
- Jiang T, Tan L, Chen Q, Tan MS, Zhou JS, Zhu XC, Lu H, Wang HF, Zhang YD, Yu JT. A rare coding variant in TREM2 increases risk for Alzheimer's disease in Han Chinese. Neurobiol Aging. 2016 Jun;42:217.e1-3. Epub 2016 Mar 3 PubMed.
- Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015 Jun;138(Pt 6):1738-55. Epub 2015 Mar 31 PubMed.
- Boissonneault V, Filali M, Lessard M, Relton J, Wong G, Rivest S. Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer's disease. Brain. 2009 Apr;132(Pt 4):1078-92. Epub 2009 Jan 17 PubMed.
- Luo J, Elwood F, Britschgi M, Villeda S, Zhang H, Ding Z, Zhu L, Alabsi H, Getachew R, Narasimhan R, Wabl R, Fainberg N, James ML, Wong G, Relton J, Gambhir SS, Pollard JW, Wyss-Coray T. Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J Exp Med. 2013 Jan 14;210(1):157-72. PubMed.
Further Reading
News
- TREM2 Data Surprise at SfN Annual Meeting
- TREM2 Tidbits at AAIC: Genetics, Clinical Data
- Alzheimer’s Risk Genes Give Up Some Secrets at SfN
- Microglial Marker TREM2 Rises in Early Alzheimer’s and on Western Diet
- Barrier Function: TREM2 Helps Microglia to Compact Amyloid Plaques
- TREM2 Buoys Microglial Disaster Relief Efforts in AD and Stroke
- Does Soluble TREM2 Rile Up Microglia?
Primary Papers
- Ulrich JD, Ulland TK, Colonna M, Holtzman DM. Elucidating the Role of TREM2 in Alzheimer's Disease. Neuron. 2017 Apr 19;94(2):237-248. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.