2018—A Year in Research
Quick Links
Once again, it’s time to reflect on the preceding 12 months. Despite the 2025 deadline for effective therapy that has been set by the National Alzheimer’s Plan and the World Dementia Council, 2018 brought no drug approvals or Phase 3 breakthroughs. Short of that, however, smaller advances happened across neurodegenerative research this past year. Therapeutic antibodies massively reduce brain amyloid—will that help? Blood tests are coming—will they draw the thousands of people needed for preclinical trials? Systems-based approaches are transforming the study of gene expression, of glial cells, and of selective vulnerability. And cryoEM is serving up eye-popping structures of tau—will they crack the FTD tauopathies and inspire new drug design? Funding is robust, raising the NIA pay line to its highest in decades. Read our yearly retrospective for highlights of 2018.
Clinical Trials
In 2018, a mix of positive and negative trial data left the field with a sense of unease that, in order to meet its goal of a game-changing treatment by 2025, everything has to go right from now on. On the up side, the SPRINT MIND trial indicated that keeping systolic blood pressure under 120 mm Hg in a person’s 60s reduced mild cognitive impairment four years later by 19 percent, while in a Phase 2B trial, the anti-Aβ protofibril immunotherapy BAN2401 seemed to both mop up Aβ plaques from the brain and slow cognitive decline in people with early AD (Aug news; Jul news). Both trials generated optimism about early intervention. The initial BAN2401 data were greeted with skepticism because of a skewed distribution of ApoE4 carriers across the treatment groups, but held up in a second analysis. (Nov news). However, no new trial has been announced yet.
BAN2401 joins the aducanumab, gantenerumab, and N3pG antibodies in removing amyloid plaques in the brain. Up to half the participants fell below the threshold for amyloid positivity over the course of one to two years (Aug news). Convinced that their early antibody efforts were too timid, researchers at Roche/Genentech and Lilly boosted dosing of gantenerumab, crenezumab, and N3pG, respectively, but as yet, none of these treatments has been shown to slow or halt dementia.

The Brain Drain You Want. Amyloid scans from three patients on gantenerumab at baseline (top row), one year (middle), two years (bottom). Reduction shown in centiloids. [Courtesy of Greg Klein.]
On the down side, the field was shaken to learn that three BACE inhibitors worsened cognition in prodromal AD. This news came after lack of efficacy had already felled Merck’s verubecestat in prodromal AD (Feb news). Verubecestat, Janssen’s atabecestat, and Lilly’s lanabecestat all weakened cognitive test performance, prompting vigorous debate at this fall’s CTAD meeting in Barcelona (Nov news). Volunteers on these inhibitors also developed more neuropsychiatric side effects than did people on placebo. The news called to mind trials of γ-secretase inhibitors, which also hastened cognitive decline. Both BACE and γ-secretase cleave dozens of substrates in addition to the amyloid precursor protein. Scientists have long voiced fears about on-target side effects, and the latest mouse studies this year did not allay those concerns since conditionally knocking out BACE1 in adults caused neuronal deficits and BACE2 turned out to function in neurons and glia (Feb news; Sep news; Mar news). Still, some scientists remain hopeful. Because the cognitive deficit did not worsen and occurred only at the highest doses, they believe a workable dose can be found. Also, they urged the field not to tar all BACE inhibitors with the same brush just yet. Novartis and Eisai/Biogen reported no cognitive worsening in their, albeit small, Phase 2 trials of BACE inhibitors CNP520 and elenbecestat, respectively. The latter blocks BACE1 more than BACE2, and seems to slow cognitive decline while reducing amyloid by 25 centiloids on PET scans. Two Phase 3 trials will test elenbecestat in prodromal AD, while two Phase 2/3 trials are currently enrolling people who carry the ApoE4 allele to evaluate CNP520 as part of the Alzheimer Prevention Initiative’s GENERATION program. One of those trials also tests the active anti-Aβ vaccine CAD106.
Taking a different tack, researchers in Spain claimed they cleared Aβ from the brain in a procedure akin to an engine oil change. They removed Aβ from blood by way of plasmapharesis, in which a person’s plasma is exchanged for a solution of 5 percent albumin, the principal carrier of Aβ in the blood, and in some cases also containing blood immunoglobulins that bind Aβ. These carrier proteins indirectly coax Aβ from the brain, the theory goes. The Phase 2b/3 AMBAR (Alzheimer’s Management by Albumin Replacement) trial missed its endpoint, but subgroup analysis suggested cognitive decline slowed in participants with moderate, though not mild, AD (Nov news). Whether the albumin or the immunoglobulins did the trick is unclear, and the blood products company Grifols plans to run a new trial to clarify. A related approach of soothing the “inflammaging” brain, either with whole plasma or defined fractions from the blood of young adults, is in early stage trials (Jul news).

Target Tau. Is catching tau by its belly (yellow) better than catching the tail (not shown)? The antibody CBTAU-24.1 (green/turquoise), which may be akin to the Phase 1 biologic JNK-63733657, binds the mid-region of tau and its aggregates in AD brain tissue [Courtesy of Zhang et al., Structure.]
In 2018, ongoing tau immunotherapy trials did not read out just yet, but researchers have been refining this approach. UCB Biopharma and Janssen reported that their therapeutic antibodies against tau’s mid-region best stop spread of proteopathic species in cell and mouse models (Apr news; Oct news); UCB00107 and JNJ-63733657 entered Phase 1 testing. AbbVie’s C2N 8E12 and Biogen’s BIIB092 bind tau’s N-terminal and are in Phase 2 for AD and progressive supranuclear palsy. The new idea is that N-terminal antibodies might prove less effective because this end of tau is exposed, gets cleaved, and contributes less to seeding than does tau’s middle, but whether this is what goes on in the human disease brain remains to be seen.
The year dished out some disappointments. Takeda’s TOMMORROW trial was found wanting. Futility analysis indicated that pioglitazone, a PPARγ agonist that improves insulin responses, wouldn’t delay progression to dementia in people who were genetically at risk due to variants around the ApoE4/Tomm 40 locus, and the trial was terminated (Jan news). Insulin itself, in an intranasal spray that has been in testing for half a decade, didn’t help people with mild cognitive impairment or mild AD in the SNIFF trial (Nov news). Likewise, nilvadipine, a calcium channel blocker, croaked in Phase 3 (Sep news). And the setbacks did not end there. In 2018, Boehringer Ingelheim’s PDE9 inhibitor BI 409306 failed, as did vTv Therapeutics’ Azeliragon, Axovant’s intepirdine and nelotanserin, and others.
With progress slow going in AD, researchers are looking to borrow from success in other neurodegenerative conditions. The U.S. Food and Drug Administration approved the first RNAi therapy, paving the way for others (Aug news). It suppresses mutant transthyretin, which causes a hereditary amyloidosis (May news). After years of study on how they regulate gene expression, antisense oligonucleotides, aka ASOs, are poised to break out. Following the 2016 approval of an ASO to treat infant spinocerebellar ataxia, researchers last spring reported that the ASO IONIS-HTT Rx halved CSF levels of mutant huntingtin in people with Huntington’s disease (Mar news), and partner Roche has begun Phase 3. Biogen and Ionis began enrolling in 2018 for a Phase 1 trial of an ASO inhibiting tau translation (BIIB080). Working preclinically, scientists created a splice-switching oligonucleotide, aka SSO. It fools the splicing machinery into omitting exon 17 when it assembles amyloid precursor protein transcripts (exon 17 contains the Aβ sequence). The SSO suppressed Aβ production when injected into mouse brain (Jun news).
Fluid Biomarkers
In 2018, the field suddenly brought what once seemed a pipe dream into reality—a blood test for AD. Ultrasensitive assays did the trick.
Hard on the heels of a first mass-spectrometry plasma Aβ assay published in 2017, independent groups early this year unveiled a second assay that reflects brain amyloid with up to 90 percent accuracy (Feb news). Soon after: a highly sensitive plasma ELISA that rivaled mass-spec approaches for sensitivity and specificity. And scientists adapted fully automated CSF assays to now work with plasma samples, as well (Aug news). Importantly, blood tests detect brain amyloid nearly as well as amyloid scans do. Hoping to avoid the high variability that plagued development of CSF assays, the field has started grappling with the tricky details of pre-analytical sample handling and assay standardization, and a round-robin distribution of a common set of plasma samples to labs testing various assay platforms is underway (Nov news).
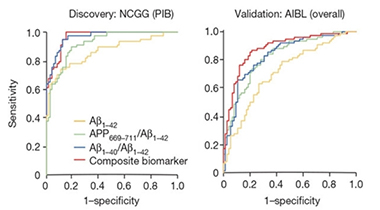
Plasma Markers. Analysis reveals high sensitivity and specificity for plasma biomarkers in Japanese (left) and Australian (right) cohorts. [Courtesy of Nakamura et al., Nature.]
Blood tau tests are around the corner, as well. Two groups described ultrasensitive assays to quantify plasma phospho-tau181, and a third published a new assay detecting N-terminal tau fragments; all distinguished controls from AD (Apr news; Dec news). This year also validated a plasma NfL test as a counterpart to the older CSF test. Longitudinal analysis correlated the rate of NfL increase in plasma with cortical thinning, solidifying the belief that plasma NfL reflects ongoing neurodegeneration (Aug news).
Brain Imaging
This year saw the culmination of the massive Imaging Dementia–Evidence for Amyloid Scanning (IDEAS) study. An effort to persuade the Centers for Medicare & Medicaid Services (CMS) to start reimbursing for amyloid PET, IDEAS was to determine whether such scans make a difference in a person’s treatment. The outcome? Among 11,409 cognitively impaired people scanned for brain amyloid, physicians indeed changed diagnoses for a third, and changed treatment plans for a majority of those. However, as it turns out, a cheaper test of Aβ in the CSF agrees with amyloid PET 90 percent of the time, as do up-and-coming blood tests (Nov news). CMS has not yet issued a decision, and IDEAS will next determine if amyloid scans make a difference to longer-term clinical outcomes.
Meanwhile, brain imaging is becoming more able to track and distinguish one neurodegenerative disorder from another. Scientists reported that Aβ accumulation is a necessary precursor for the broad invasion of the cortex by tau aggregates that is a mark of Alzheimer’s, and that this invasion coincides with cognitive impairment in people with sporadic and familial AD (Feb news; Feb news). Another familial AD study pieced together a cascade starting with Aβ accumulating in the precuneus, followed by waning metabolism, then atrophy (Feb news).

Amyloid to Atrophy. The first signs of amyloid, hypometabolism, and cortical thinning by marker and by brain region. [Courtesy of Gordon et al., Lancet Neurology, 2018.]
The first published autopsy report of a patient who had been scanned twice with the tau PET tracer flortaucipir showed that ligand uptake tightly correlated with tau pathology across wide swaths of the brain (Dec news; Nov news). Flortaucipir, which binds the type of tangle found in AD, is not useful for non-AD tauopathies, and other tau tracers are pressing in to fill the space (Sep news; May news).

Life and Death. In a man with early onset AD, uptake of flortaucipir in life matched with postmortem AT8 staining in neurites, soma, or overall, in 15 brain regions. [© 2018 American Medical Association. All rights reserved.]

Tau PET Sees AD Dementia. AD patients (left) accumulate tau tangles across large swaths of the brain (color shows intensity versus control); this signal is much lower at the MCI stage (right). [Courtesy of Ossenkoppele et al., ©2018, American Medical Association. All rights reserved.]
Besides Aβ and tau, scientists want to track many other targets in the aging brain, and they are calling for PET ligands. The year 2018 came up short on α-synuclein or TDP-43, but brought some progress on imaging synapses and epigenetic modification. A group of scientists continued to build their case for UCB-J. A derivative of the anti-epileptic drug levetiracetam, this ligand binds synaptic vesicle glycoprotein 2A, essentially quantifying synaptic density. PET scans showed less binding in the hippocampus of AD than control, a pattern that correlated with memory (Aug news). And in a surprise, a study to assess histone deacetylase enzymes by PET with the tracer Martinostat found that class I HDACs drop as AD advances, rather than increase, as people had predicted based on prior animal data (Aug news).
A Diagnosis Rooted in Biology, Not Symptoms
Against the backdrop of new biomarker development, a committee assembled by the National Institute on Aging and the Alzheimer’s Association in 2018 proposed a new research classification for the diagnosis of AD in an effort to nudge the field toward a biological definition of the disease (Apr news). The AT(N) framework makes the diagnosis of AD when there is biomarker evidence for amyloid plaques (A) and neurofibrillary tangles (T), whether or not a person shows symptoms. The extent of neurodegeneration (N) helps stage the disease. NfL has emerged as a strong contender for the N of AT(N) (Nov news).
This biological definition continues a shift away from the syndromic, clinical diagnosis based on cognitive changes such as memory loss, confusion, or dementia. The shift began in 2007 and picked up steam in 2012, when NIA/AA postmortem diagnosis criteria were updated to focus exclusively on neuropathology, regardless of whether the diseased person had shown symptoms of AD.
The 2018 diagnostic framework committee also outlined a symptom-based staging scheme for AD that parallels biomarker profiling, with six stages that cover the spectrum from cognitively normal to severely demented. Researchers are now probing this new framework in their longitudinal research cohorts, and early indications are that biomarkers for amyloid, tau, and neurodegeneration, when used in combination, do a good job of predicting a person’s progression (Nov news). In the process, scientists are also refining the field’s familiar biomarker staging diagram, and amending it with new markers, such as sTrem2, NfL, progranulin, and YKL-40 (e.g. Nov news). The field urgently needs better cognitive measures to define the clinical stages, especially early on.
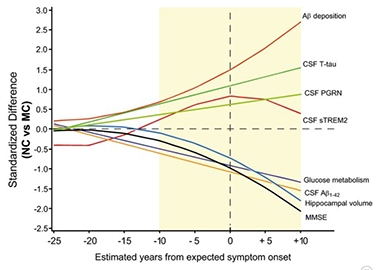
Progranulin in Context. In this ADAD staging diagram, CSF progranulin creeps up about 10 years before a mutation carrier is expected to develop dementia. [Courtesy of Suárez-Calvet et al., 2018.]
The FDA joined this collective push toward putting AD research on a biological footing. In March, the agency issued new guidance on clinical trials for pre-dementia. It recognizes three stages: In stage 1, only biomarkers are abnormal; in stage 2, subtle cognitive effects crop up, too; and in stage 3, functional deficits. In a shift, the agency said it will consider approving drugs based solely on neuropsychological test scores in preclinical disease. (Mar news).
Trials at this early stage require preclinical cohorts of people whose genetic risk and biomarker status are known. This is a big challenge for the field, and 2018 was spent by groups around the globe preparing for such trial-ready cohorts (TRCs) in their respective countries. For example, the European Prevention of Alzheimer’s Dementia (EPAD-TRC) and the U.S. Preclinical/Prodromal Alzheimer’s Disease (TRC-PAD) are hoping to build standing trial platforms. Both are off to a slow start. The EPAD-TRC had recruited 1,111 volunteers as of October 2018, far shy of its original goal of 6,000, which has been revised down. The TRC-PAD thus far has recruited 7,400 of a planned 50,000 people into an online feeder study; in-person screening will start in 2019 (Nov news).
In October, the NIA issued a national strategy document to boost representation of minorities in dementia research. Also, stakeholders gathered to debate how best to boost participation of black Americans in Alzheimer’s studies specifically. Like Latino Americans, African-Americans are disproportionately affected by dementia, yet are underrepresented in observation studies and clinical trials (Jul news).
Genetics
In 2018, researchers upped the ante on genome-wide association studies (GWAS) to dig up additional polymorphisms linked to AD risk. Some studies piled on more participants to classic GWAS, while others used a technique dubbed GWAX, in which family members of people with AD are counted among the cases. In all, 13 new risk loci emerged in 2018, bringing the total number of common AD risk hits above 30 (Apr news). Researchers also made strides linking GWAS hits, many of which reside within noncoding regions of the genome, to the nearby genes that might functionally cause the risk. For example, techniques to catch enhancers in regulatory liaison with genes they control tied GWAS hits to specific genes, and in some cases the gene was not the one nearest the polymorphism. Researchers also took nuclei from different cell types in the brain to determine where such regulatory relationships played out. Often, it was in microglia (Jul news).
In the hunt for rare variants with strong effects, geneticists are sequencing whole exomes and, in August, announced results of the largest WES analysis to date. Sorting through 20,000 exomes in the Alzheimer’s Disease Sequencing Project, researchers pulled out but two hits, one in a zinc finger protein and the other in a transcription factor. A variant in an immunoglobulin that binds Aβ missed statistical significance (Aug news).
Advances in mouse transcriptomics enabled RNA-Seq to measure expression changes as microglia responded to the build-up of amyloid plaques. This uncovered a network of microglial genes, including five potentially new AD culprits. Their human orthologues lie near single-nucleotide polymorphisms that associate with the disease (Dec news). Researchers hope that transcriptomics combined with network analysis will define disease pathways.
Immune system
Interest in microglia picked up well before 2018, when AD risk variants kept cropping up near microglial genes, not least the receptor TREM2. This year deepened scientists’ understanding of how TREM2 protects the brain, and also broadened their appreciation of just how dynamic and manifold microglial subtypes are. Researchers reported that Aβ binds TREM2, activating microglia and setting off a signaling cascade that spurs them to degrade the peptide. TREM2 makes microglia less inflammatory and more phagocytic, reducing amyloid in mice (Mar news). And without TREM2, Aβ plaques form more readily, are larger, less dense, and have twice as much ApoE (Jul news).
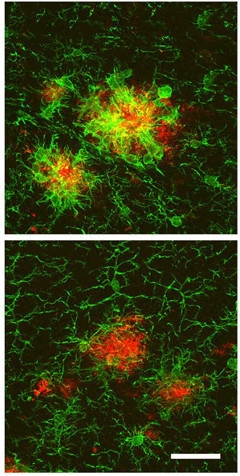
TREM2 Remodels Microglia. In AD mice (top), microglia surrounding plaques (red) become ameboid and express high Iba1 (green). When TREM2 levels are high (bottom), microglia have longer processes and express less Iba1. [Courtesy of Neuron, Lee et al.]
Researchers homed in on TREM2’s R47H variant, which triples the risk of AD. January saw a report that soluble TREM2, the fragment cleaved by γ-secretase, latches onto plaques and neurons, whereas the soluble R47H variant has difficulty doing so (Jan news). A warning shot came when R47H turned out to have profound effects in mice that are absent in people. In particular, it scuppers splicing only of the mouse gene and completely suppresses expression, implying that mice expressing R47H TREM2 are prone to artifacts (Sep news).
As a first step toward understanding how genetic variants affect microglial function, researchers in 2018 embraced single-cell transcriptomics and proteomics to start characterizing microglial subtypes. One study of nearly 80,000 microglia from mice across the age span identified at least nine distinct subtypes that wax and wane during development, aging, or after injury (Dec news). Similar work is being done with human microglia. To avoid confounds of postmortem tissue, researchers turned to fresh autopsy or biopsy samples for single-cell analyses. This identified 14 subclusters with unique gene-expression profiles, though the sample was too small to link phenotypes to disease (Jul news). Some subtypes were enriched in genes linked to Aβ, tau, and α-synuclein. All microglial types expressed TSPO, suggesting PET ligands for this receptor may not distinguish subtypes. A proteomics study identified four microglial subtypes residing in different regions of the human brain (Dec news). While the field is further behind in characterizing astrocytes, they, too, seem to change their transcriptomes, adopting a pathogenic “A1” phenotype as mice age (Feb news). And in a reminder that immunity in the brain remains a wild frontier of research, scientists discovered secret passageways connecting bone marrow in the skull directly to the parenchyma. The tiny corridors provide an express route for marrow cells, including neutrophils, to cross into the cortex in response to injury. The finding challenges the prevailing view in the field that bone marrow-derived cells invading the brain hail from the blood, which carries cells collectively released from all other bone marrow in the body (Aug news).
Viruses and Transposons
New research in 2018 revived interest in microbes and dementia, a link that has been documented for decades but widely disbelieved. In an unbiased, multi-cohort, multi-network analysis of AD brain, researchers turned up herpes and other viral sequences (Jun news). Analysis of data in the AMP-AD project tied viral expression to AD but could not say whether the viruses cause the disease or merely take advantage of weakened cells to flex their genomes.

Viruses and AD. Multi-omic evidence links viral load to the molecular, pathological, and clinical manifestations of dementia. [Courtesy of Ben Readhead.]

Upregulated Host Genes. Human genes upregulated by herpes virus 6A include PSEN1, CLU, BIN1, and PICALM. [Courtesy of Readhead et al., Neuron 2018.]
Either way, researchers see important ramifications. Nearly half of the human genome consists of transposable elements, including ancient relics of viruses that hitched a ride during evolution. Most of these are silent, but this year researchers reported that tau pathology opened chromatin around retrotransposons, leading to their aberrant expression in neurons from AD brain and in fly models of the disease (Jun news; Jul news). On the other hand, herpes viruses were reported to seed aggregation of Aβ, which purportedly ensnared and neutralized them, protecting cells and mice (Jun news).
Proteotoxicity
Scientists turned up further molecular proof that distinct neurodegenerative diseases are caused by specific conformations, or strains, of proteins. The structure of aggregated tau isolated from the brain of a person with Pick’s disease assumed a J-shaped conformation, which looked quite different from the C-shaped fold of Alzheimer’s tau reported the year before (Aug news).

Different Strokes. In Pick’s disease (left), tau molecules adopt a long J-shaped fold, while in AD (right) they form a C that contains fewer domains. [Courtesy of Falcon et al., Nature.]
Single Tau Molecule. CryoEM of a wide tau filament from a Pick’s disease patient shows two tau J folds attached at their tips. [Courtesy of Falcon et al., Nature.]

Regarding Aβ, it turned out that only a tiny fraction of its species in the brain is toxic (Jul news). This could affect treatment if therapeutic antibodies get waylaid by inactive forms of the peptide. While the toxic species may be sparse, they seem extremely stable. After 30 to 40 years of storage as a powder at room temperature, extracts from human pituitary glands seeded brain amyloid plaques in transgenic mice expressing human APP. The work supports the idea that Aβ contamination caused the amyloid pathology detected in young adults who received growth hormone treatment as children (Dec news).
Researchers claimed that these cadaver preparations also transmitted tau and, separately, that Aβ transferred into the brain during neurosurgery caused early and devastating cerebral amyloid angiopathy in a handful of patients (Feb news; Duyckaerts et al., 2018).

Sidelined. Unlike Aβ fibrils that hold tightly together by interactions between side chains (top panel), LARKS fibrils (bottom) are weakened by a kink that limits their interface. [Courtesy of Science/AAAS.]
How do toxic proteins arise in the brain? This year saw more evidence that proteins with low-complexity domains undergo liquid-liquid phase separation, forming dense liquid droplets within cells that may be hotbeds for formation of amyloids (Feb news). The thrust of this research currently is around C9ORF72 and other proteins implicated in ALS/FTD. In the case of toxic poly-dipeptide repeats encoded by hexanucleotide expansions in intron 1 of the C9ORF72 gene, cell stress seems to be a driver. At the beginning of 2018, scientists discovered that stress promotes RAN translation, whereby the expansions are bidirectionally transcribed to generate five different poly-dipeptide repeats (Jan news). By the end of the year, scientists also knew that repeat-containing transcripts themselves triggered the very stress response that ramped up RAN translation, while loss of C9ORF72 function caused by the expansion hobbled autophagy, exacerbating dipeptide repeat toxicity (Nov news). Cell type may also be a factor in toxic protein formation. For example, aggregates of α-synuclein that formed in, or passed through, oligodendrocytes turned out to be much worse than those that arose in neurons (May news), while excitatory neurons seemed more susceptible to tau toxicity than inhibitory ones because the latter were better at clearing unwanted tau by autophagy (Dec news).

Tau’s Tangled Web. Groups of co-expressed genes associated with protein aggregation (red), tau tangles (green), aggregation protectors (blue) and promoters (yellow, upper left), tau (black), and genes shared between MS and tangles (brown). [Image courtesy Fu et al., Nature Neuroscience.]
Similarly, in the ongoing push to find out how toxic proteins spread through the brain, hints came from studying tau. One group reported that hyperphosphorylated tau latched onto synaptic vesicles, another that neuronal activity coaxed tau into presynaptic terminals, whereupon microglia engulfed tau-laden vesicles, damaging synapses and unwittingly spreading toxic tau (Feb news; Jul news).
And for the old culprit, Aβ, 2018 upended the widely held notion that amyloid plaques can accumulate for years in a person’s brain before causing cognitive symptoms. More sensitive memory tests now detect slippage in test scores in people who are just beginning to deposit plaques, but are still below the threshold for brain-wide positivity, compared with people who are not depositing amyloid (Aug news).
Sleep on It
Soaked it all in yet? Data suggests you should certainly sleep on it. This year, scientists strengthened the connection between sleep deprivation and amyloid pathology. A single night without sleep was said to boost Aβ production by 30 percent, and excessive daytime sleepiness over two years led to faster amyloid deposition in healthy older people (Jan news; Mar news). The sleep-amyloid relationship may be a two-way street. Amyloid precipitates circadian disturbances in adults, researchers claimed, while damage to the endogenous circadian clock in transgenic mice brought on more plaques, activated microglia, and runaway astrogliosis (Feb news; Dec news).
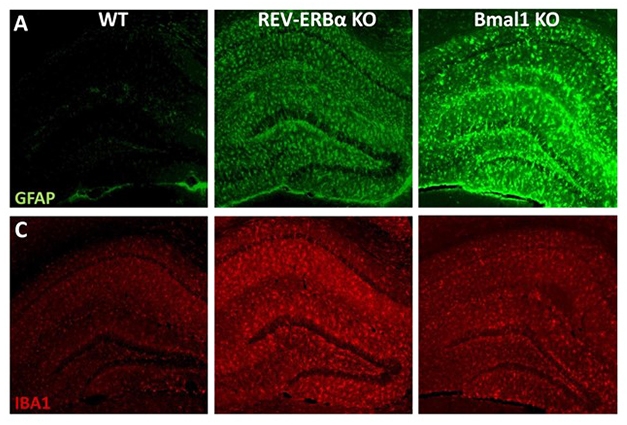
Out of Sync. Knocking out REV-ERBα or Bmal1, central components of the circadian oscillator, activates microglia (red, Iba1) and to a lesser extent, astrocytes (green, GFAP). [Courtesy of Erik Musiek.]
If clarity about any of this does not come with sleep, then blame it on the season. No, it is not necessarily too much partying. According to one study, people seem to be at their cognitive peak in late summer and then trough early in the new year, at least in the northern hemisphere (Sep news).
Funding
Two thousand eighteen brought good news on the money front. In August, Congress passed a 2019 budget that would increase spending on AD research by $425 million, bringing it up to $2.34 billion (Aug news), and President Trump signed the bill into law in September. A fiscal year 2019 appropriation of $3.08 billion enabled the National Institutes on Aging to boost its 2019 pay lines for Alzheimer’s and related disorders grants into the 25 to 38 percent range (NIA FY2019), a far cry from a few years ago, when they dipped to 9 percent (Mar 2011 news). In February, the Chan Zuckerberg Initiative, part of a larger $3 billion philanthropy, announced that the CZI Neurodegeneration Challenge Network would support basic research in neurodegeneration, and in December it awarded a first set of 26 grants totaling $51.95 million to support early career investigators and small collaborative cross-disciplinary teams (Feb news; Dec news). July saw the announcement of the Diagnostics Accelerator, which will fund commercial development of diagnostic biomarkers for early AD with $30 million of investing from Bill Gates, the Alzheimer’s Drug Discovery Foundation, and others (Jul news). And earlier this month at the World Dementia Council summit in London, the U.K. government pledged up to ₤79 million to establish a 5 million-strong, research-ready cohort that can be screened by artificial intelligence to improve detection, diagnosis, and treatment of AD and other dementias (Dec news).—The Alzforum News Team.
References
News Citations
- Could Better Blood Pressure Management Preserve Cognition?
- BAN2401 Removes Brain Amyloid, Possibly Slows Cognitive Decline
- Second Look at BAN2401 Data Still Positive, Despite Snafu
- Four Immunotherapies Now Banish Amyloid From the Brain
- Merck Axes Verubecestat for Prodromal AD, Researchers Say ‘Go Earlier’
- Bump in the Road or Disaster? BACE Inhibitors Worsen Cognition
- BACE1 Conditional Knockouts Model Adult BACE Inhibition
- In Conditional BACE1 Knockouts, Hippocampal Axons Compromised
- Remember: BACE2 Is in the Brain, Too
- Fits and Starts: Trial Results from the CTAD Conference
- VCAM1: Gateway to the Aging Brain?
- To Block Tau’s Proteopathic Spread, Antibody Must Attack its Mid-Region
- New Antibody Binds to Mid-Region, and Aggregates, of Tau
- There’s No Tomorrow for TOMMORROW
- Trials of Diabetes-Related Therapies: Mainly a Bust
- Nilvadipine Fails to Slow Cognitive Decline in AD Patients
- FDA Approves First RNAi Therapy
- As RNA Therapies Come of Age, Efficacy Remains Weak
- Antisense Therapy Cuts Huntingtin Protein in CSF by Half
- Antisense Oligonucleotides Turn Down the Aβ Spigot
- Closing in on a Blood Test for Alzheimer’s?
- With Sudden Progress, Blood Aβ Rivals PET at Detecting Amyloid
- Blood Tests for Amyloid Step Out at CTAD
- After Plasma Aβ, Now Plasma P-Tau181 Shows Promise
- Could a Blood Test for Tau Diagnose Alzheimer’s Disease?
- Blood Test for Neurofilament Light Chain Kicks Up Biomarker Research
- Amyloid PET Aids Diagnosis. But Could CSF Do Just as Well?
- Imaging Clinches Causal Connections between Aβ, Tau, Circuitry, and Cognition
- In Familial Alzheimer’s, Tau Creeps into Cortex as Symptoms Show
- Aβ, Then Metabolism, Then Atrophy: In Familial AD, Cascade is Definitive
- Autopsy Study Confirms Flortaucipir PET Lights Up Tau Pathology
- It’s Official: Tau PET Sees Tangles, and Staging Tangles Predicts Decline
- Flortaucipir Imaging Distinguishes Alzheimer’s From Other Disorders
- Move Over, Flortaucipir? New Tau Tracers Tested in People
- PET Ligand Lights Up AAIC, May Detect Synapse Loss in AD
- Surprise: HDAC Goes Down, Not Up, in Alzheimer’s Disease
- New Definition of Alzheimer’s Hinges on Biology, Not Symptoms
- Is NfL the New 'N' in A/T/N Classification of Alzheimer’s?
- How Does The NIA-AA Framework Measure Up Against Real Data?
- Progranulin: A Mark of Worsening Alzheimer’s?
- FDA OKs Cognition as Sole Outcome Measure for Preclinical AD Trials
- Trial-Ready Cohorts Take Shape in Europe and U.S.
- Alzheimer’s Researchers Seek Advice on How to Include African-Americans
- GWAS, GWAX: bioRχiv Hosts Bonanza of Alzheimer’s Genetics
- A Delicate Frontier: Human Microglia Focus of Attention at Keystone
- Largest AD Whole-Exome Study to Date Finds Two New Risk Genes
- Microglia Reveal Formidable Complexity, Deep Culpability in AD
- TREM2 Binds Aβ, Reprograms Microglia to Curb Plaques
- TREM2: Diehard Microglial Supporter, Consequences Be DAMed
- New Mouse Models Reveal Unexpected Property of TREM2
- Model Morass? R47H Mutation Scuttles TREM2 Expression in Mice, Not People
- Local Flavor: At Protein Level, Too, Human Microglia Are Diverse
- Aged Astrocytes Prime Brain for Neuroinflammation
- Tiny Passageways Connect Skull Bone Marrow to the Brain
- Aberrant Networks in Alzheimer’s Tied to Herpes Viruses
- Tau Aggregates Awaken Genetic Relics in the Brain
- Jumping Genes Rampant in Tau Flies
- Herpes Triggers Amyloid—Could This Virus Fuel Alzheimer’s?
- Conformers Confirmed: Structure of Pick’s Tau Distinct from AD Tau
- A Minority of Human Aβ Species are Toxic, Good Drug Targets
- Confirmed: Human Pituitary Extract Linked to Amyloidosis Contains Aβ Seeds
- Can Aβ Seeds Be Transferred During Neurosurgery?
- Are Reversible Amyloids Behind Liquid-Liquid Phase Separation?
- Stressed-Out Cells Translate C9ORF72 Repeats, Unleash Toxic Peptides
- It’s ‘And,’ Not ‘Either-Or’: C9ORF72 Mechanisms of Action are Linked
- Oligodendrocytes Seem to Imprint Specific Alter Ego on α-Synuclein
- Tau Silences, Aβ Inflames; Hitting Excitatory Synapses Hardest
- Tau Uses Synaptogyrin-3 to Clump Synaptic Vesicles
- Synaptic Tau Clangs the Dinner Bell for Hungry Microglia
- Memory Slips as Soon as Amyloid Appears, Two Decades Before Dementia
- Skimping on Sleep Makes For More Aβ in the Brain
- Does Daytime Drowsiness Foreshadow Aβ Accumulation?
- Faulty Brain Clocks Precede Alzheimer’s, May Drive Pathology
- When Glial Clocks Fall Out of Sync, Inflammation Ensues
- Does Cognition Change With the Seasons?
- Senate Approves $2.34 Billion Budget for Alzheimer’s Research
- Funding Woes: What’s Behind the Falling NIA Payline?
- Chan Zuckerberg Initiative Seeks Collaborators for Basic Research in Neurodegeneration
- First Round of Awards from Chan Zuckerberg Initiative Announced
- $30 Million Venture Philanthropy Project for AD Biomarkers
- World Dementia Council Takes Stock of Progress, Challenges
Therapeutics Citations
- Umibecestat
- Elenbecestat
- Amilomotide
- Tilavonemab
- Gosuranemab
- BI 409306
- Azeliragon
- Intepirdine
- Nelotanserin
- BIIB080
- Levetiracetam
Paper Citations
- Duyckaerts C, Sazdovitch V, Ando K, Seilhean D, Privat N, Yilmaz Z, Peckeu L, Amar E, Comoy E, Maceski A, Lehmann S, Brion JP, Brandel JP, Haïk S. Neuropathology of iatrogenic Creutzfeldt-Jakob disease and immunoassay of French cadaver-sourced growth hormone batches suggest possible transmission of tauopathy and long incubation periods for the transmission of Abeta pathology. Acta Neuropathol. 2018 Feb;135(2):201-212. Epub 2017 Nov 22 PubMed.
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.