In Alzheimer Brain, Can Synaptic Pruning Be Good?
Quick Links
In the Alzheimer’s disease brain, synaptic loss correlates with cognitive decline, and is considered a sign of disease progression. But is synaptic loss always bad? Provocative new data from several groups suggest that early in disease, selective glial pruning of synapses could protect the brain by calming overactive neuronal circuitry. The microglial receptor TREM2 mediates this, allowing microglia to recognize and snip hyperactive connections.
- In the presence of Aβ, hyperactive synapses get tagged with phosphatidylserine.
- Microglial TREM2 recognizes this tag and prunes the synapses.
- This cools network excitability, protecting the brain.
What’s the evidence? In the August 15 Neurobiology of Disease, scientists led by Lennart Mucke at the Gladstone Institute of Neurological Disease, San Francisco, reported that amyloidosis mice carrying the hypofunctional TREM2 variant R47H developed epileptiform discharges as they aged, along with an excess of cortical synapses. In parallel, researchers led by Soyon Hong at University College London delineated a potential underlying mechanism. In the August 14 EMBO Journal, they showed that Aβ oligomers caused synapses to become overactive and express the “eat me” signal phosphatidylserine. Microglia snipped these spines away, restoring normal activity—but not if the cells expressed R47H.
In the August 25 Cell Reports Medicine, researchers led by Tara Spires-Jones and Barry McColl at the University of Edinburgh extended the findings to people, showing that astrocytes and microglia gobble more synapses in AD than control brain. As in mice, this is mediated by phosphatidylserine, along with a binding partner, MFG-E8.
“Together, these papers tell us that synapse elimination is not necessarily bad,” Michela Matteoli at Humanitas University in Milan, Italy, told Alzforum (see Q&A below). She noted that whether synaptic pruning helps or harms the brain depends on context. “The key point is likely the time window, and the nature of the synapses eliminated,” she suggested.
Pruning in Action. A cultured microglia (yellow) extends a process toward a synapse marked with phosphatidylserine (pink) and engulfs it, but ignores unmarked synapses (green). [Courtesy of Rueda-Carrasco et al., EMBO Journal.]
Mucke and others had previously linked Aβ to overactive neuronal circuitry at early disease stages in both mice and people (May 2012 news; Aug 2012 news; May 2012 news). Two years ago, Mucke’s group reported that lowering TREM2 in mice worsened epileptiform activity (Das et al., 2021).
The scientists followed up on this by examining mice that carry the human R47H TREM2 variant, which is associated with a threefold higher risk of AD. First author Melanie Das challenged the R47H transgenics with kainic acid, which induces epileptic activity. The transgenics were more susceptible than were controls, experiencing twice as many electrical spikes. When crossed with APP knock-in mice, the transgenics had three times as many cortical electrical discharges after one year of age as did APP mice with normal TREM2. The R47H-APP mice had about 50 percent more cortical synapses than did littermates with normal TREM2.
Hong and colleagues tied hyperactivity to microglial engulfment. Joint first authors Javier Rueda-Carrasco, Dimitra Sokolova, and Sang-Eun Lee added synthetic Aβ oligomers to mouse hippocampal neurons. This ramped up neuronal activity, as seen by calcium imaging. The most active dendritic spines flipped the membrane lipid phosphatidylserine from the inner to the outer layer, exposing it on the cell surface. External phosphatidylserine (ePtdSer) has previously been shown to act as an “eat me” signal for apoptotic neurons, as well as for unwanted synapses in the developing brain (Aug 2018 news; Scott-Hewitt et al., 2020; Li et al., 2020).
In keeping with this, when mouse microglia were added to the cultures, they preferentially engulfed ePtdSer-expressing synapses. Within 48 hours, neuronal activity dropped back to baseline. When the scientists blocked ePtdSer, neurons stayed hyperactive (image below). Likewise, when microglia expressing R47H TREM2 were added, they were unable to prune ePtdSer-expressing synapses.
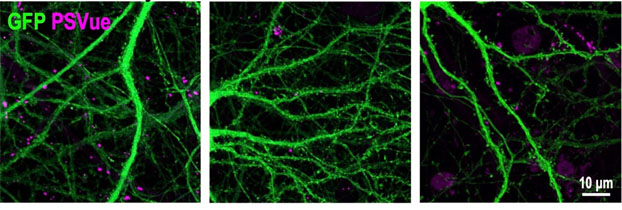
Key Signal. Mouse neurons (green, left) exposed to Aβ oligomers express phosphatidylserine (pink) at synapses. With microglia there (middle), these synapses are eliminated—but not if the phosphatidylserine signal is blocked (right). [Courtesy of Rueda-Carrasco et al., EMBO Journal.]
A similar dynamic played out in vivo. When the scientists crossed R47H knock-in mice with APP knock-ins, the R47H offspring had three times as many ePtdSer synapses as did their littermates with normal TREM2. Their microglia contained half as much synaptic material.
Youtong Huang, in Beth Stevens’ group at the Broad Institute of MIT and Harvard, noted that the paper is the first to show that ePtdSer can mediate phagocytosis of small structures such as synapses in the adult brain. Others had reported the lipid labels whole cells for removal (Segawa et al., 2014).
What about human brain? Spires-Jones and colleagues addressed this. Joint first authors Makis Tzioras and Michael Daniels analyzed postmortem tissue samples from the inferior temporal lobes and visual cortices of 32 AD brains at Braak stage 5 or 6 and 18 age-matched controls. They found twice as much synaptic material inside microglia and astrocytes in AD brains. This is the first direct evidence that glia in AD brains eat synapses, as they do in amyloidosis mice, Spires-Jones told Alzforum.
Curiously, astrocytes contained about 10 times as much synaptic material by volume as did microglia, suggesting they might be the primary phagocytes in human brain. Synapse engulfment tended to occur near amyloid plaques, and was more rampant in APOE4 than APOE3 carriers. Alas, the study was unable to determine if glia were eliminating aberrant or healthy synapses. Spires-Jones plans to study this in organotypic human brain slices from surgical resections, in collaboration with Claire Durrant at Edinburgh.
To learn how the pruning might work, the authors isolated synaptosomes from AD and control brain samples, then added them to cultured human astrocytes or microglia. Both types of glia gobbled up AD synaptosomes faster, and more of them (image below). Here microglia outdid astrocytes, cleaning up synaptosomes within an hour compared with two days for astrocytes.
What made AD synaptosomes so tasty? The authors laid the blame on MFG-E8, an opsonin produced by glia that binds ePtdSer and helps glial receptors recognize it. Opsonins bind molecules, such as antibody-antigen complexes, and make them easier to phagocytose. AD synaptosomes contained twice as much MFG-E8 as control ones, and when this was blocked by an antibody, glial appetites waned. A small fragment of MFG-E8, known as medin, accumulates as an amyloid in blood vessel walls and has been linked to cerebral amyloid angiopathy (Nov 2022 news).
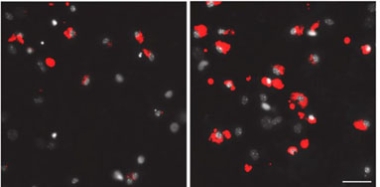
Tastier Morsels. Cultured mouse microglia (nuclei are gray) eat many more synapses (red) from AD (right) than control brain (left). [Courtesy of Tzioras et al., Cell Reports Medicine.]
Altogether, Mucke believes these papers imply that network hyperexcitability and immune cell dysfunction form a vicious circle in Alzheimer’s disease, each exacerbating the other. For example, inheriting the R47H variant would make microglia less able to contain hyperexcitability caused by a buildup of Ab oligomers with age, and the resulting network dysfunction could then worsen inflammation. “One can enter, or block, this vicious cycle from either branch,” he suggested.
More research is needed before scientists can define therapeutic targets associated with synaptic pruning. They need to know what factors determine whether synapse elimination is helpful or harmful. Likely, disease stage and brain region will matter. R47H TREM2 APP mice were losing hippocampal synapses at the same time as they were accumulating cortical ones. This suggests that pathological pruning occurs in the brain at the same time as beneficial pruning, but in different regions, Mucke noted.
How to distinguish aberrant from healthy synapses? ePtdSer and MFG-E8 provide clues but are useless for therapeutic interventions, the scientists said. Spires-Jones noted that MFG-E8 plays important roles in the periphery and protects neurons in models of stroke, hence globally suppressing it could be harmful. She is investigating other ways to target the mechanism, perhaps by blocking MFG-E8 binding to astrocytic receptors.
Beyond potential therapeutic angles, the new data help illuminate the pathogenesis of AD. “What is really exciting about these papers is they suggest that aberrant neuronal activity is one of the earliest things to go wrong in vulnerable brain areas,” Stevens told Alzforum. “We know activity is a critically important signal that regulates synapse pruning in development. These papers help connect the dots between upstream signals and phagocytosis.” Stevens is a co-author on Hong’s paper.—Madolyn Bowman Rogers
Q&A with Michela Matteoli, Humanitas Clinical and Research Center, Milan, Italy.
Questions by Madolyn Bowman
Q: Two of these papers imply that synapse elimination early in AD could be "good" by restoring normal neuronal excitability. This seems at odds with prior studies linking synapse loss to cognitive decline. What might explain the discrepancy?
A: The Rueda-Carrasco and the Das papers, building upon previous evidence, make a truly innovative and interesting point. Five years ago, my group had demonstrated that TREM2 is required for proper supernumerary synapse elimination during brain development (Filipello et al., 2018) and later that externalization of PtdSer at synapses is required for this process to occur (Scott-Hewitt et al., 2020).
The group of Soyon Hong has now investigated the same process in the context of AD. They demonstrate that Aβ oligomers induce enhanced calcium activity, which drives synaptic exposure of PtdSer and consequent synapse elimination by microglial TREM2. This process results in amelioration of neuronal hyperactivity. In parallel, the Mucke group demonstrates that the R47H, nonfunctional variant of human TREM2 (a risk factor for AD), impairs the ability of microglia to suppress network hyperexcitability caused either acutely, by injection of low doses of kainic acid, or chronically, by expression of FAD-mutant APP/Aβ. Thus, both papers underline a key role of microglia in suppressing network hyperactivity in the AD context.
Interestingly, both papers, in a direct (Rueda Carrasco) or indirect (Das) manner, identify the process of synapse elimination as the key process controlled by TREM2 even in AD. Consistently, the Das study shows that the defective TREM R47H mouse does not display increased overall plaque burden, but rather shows a significant increase in the density of cortical synapses. Thus synapses, more than plaques, are on the stage at the early stages of the disease.
Why should this synapse elimination be “good” for brain health? Network hyperexcitability is an early sign in AD, and several studies on patients have shown that AD is associated with “subclinical” non-convulsive epileptiform activity, which results in faster cognitive decline. Therefore, the elimination of hyperactive synapses may counteract epileptogenesis and prevent the establishment of maladaptive circuits that, in turn, could promote further network hyperexcitability and eventually neuronal death.
The third paper, by Tzioras and colleagues, illustrates a rather different scenario: it shows that microglia and astrocytes in human AD brains contain higher amounts of synaptic proteins than in control brains, and that in vitro these glial cells preferentially ingest AD-derived synapses compared to controls. This study is more in line with the generally accepted concept that excessive, abnormal, synapse elimination occurs in AD.
The two views are not at all in contrast, the key point being the time window and, most likely, the nature of synapses eliminated. While Rueda Carrasco and Das focus on early stages of AD, Tzioras analyzes brains of subjects with late-stage disease. It is conceivable that the same process of synapse elimination may be harmful at later stages of AD, being instead favorable at early stages of the disease. Also, the synapses shown to be eliminated in the Rueda Carrasco paper are dysfunctional, hyperactive synapses. The nature of synaptic material engulfed in the human AD astrocytes and microglia investigated by Tzioras is not defined, at least at this stage. It is likely that while the elimination of dysfunctional synapses may be good, the elimination of functional synapses is instead deleterious.
Q: Might these findings affect therapeutic approaches to AD? Will researchers have to be careful using approaches such as blocking complement, which repress synapse pruning in general?
A: Yes, this is a crucial point to be taken into account. On one side, we expect that therapeutic approaches potentiating synapse elimination will be bad; at the same time we also think drugs counteracting synapse elimination will not solve the AD issue. Indeed, while clearing dysfunctional or dead synapses could be beneficial, removing functional synapses could be harmful in contributing to synaptic degeneration. Both processes are likely to occur at different stages of disease.
Thus, the time window when we may think to exploit a specific approach is a first point to consider. The second one is that, despite advancements in the field, we need a clearer understanding of glia-synapse interactions to develop effective therapeutics to protect synapse function. We do not expect that a single mechanism, e.g., the one involving TREM2 and PtdSer, is solely responsible for synapse elimination. It is much more likely that several, concomitant actors are involved. If we were able to discover a specificity in the molecular components involved in the elimination of dysfunctional versus functional synapses, we could have in our hands potentially successful targets.
Q: The third paper finds a role for MFG-E8 in phagocytosis of synapses. Could that be a therapeutic target?
A: In principle, yes, just like the integrin receptor α5β5, which binds MFG-E8, as well as C1q, or all the other molecules involved in synapse elimination. The real problem is the point above. If we univocally demonstrate that MFG-E8 (or other components) comes into play specifically in the pathological elimination of functional synapses, then this would be a good starting point to think about it (or them) as possible therapeutic target.
Q: Overall, how do these three papers expand our understanding of the role of glial-mediated synapse loss in AD?
A: The three papers tell us a lot more about glial-mediated synapse loss in AD. They add a layer of complexity to the scenario. Together with evidence from previous studies, these papers tell us more clearly that synapse elimination is not necessarily bad. The process of synapse elimination is specifically required to direct formation of the correct brain circuit (see for example our previous papers) as well as to suppress unwanted network excitability (Rueda Carrasco and Das studies). That said, generalized strategies to impaire synapse elimination as a therapeutic target for AD would likely be a complete failure.
References
News Citations
- Soluble Aβ Takes Blame for Hyperactive Neurons in Mouse Brain
- Anticonvulsants Reverse AD-like Symptoms in Transgenic Mice
- Epilepsy Drug Calms the Hippocampus, Aids Memory
- Tangles Turn Neuronal Membranes Inside Out, Give Microglia License to Eat Their Fill
- Meddling Medin: A Vascular Amyloid That Promotes CAA?
Research Models Citations
Paper Citations
- Das M, Mao W, Shao E, Tamhankar S, Yu GQ, Yu X, Ho K, Wang X, Wang J, Mucke L. Interdependence of neural network dysfunction and microglial alterations in Alzheimer's disease-related models. iScience. 2021 Nov 19;24(11):103245. Epub 2021 Oct 7 PubMed.
- Scott-Hewitt N, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, Carey A, Faggiani E, Schuetz LT, Mason S, Tamborini M, Bizzotto M, Passoni L, Filipello F, Jahn R, Stevens B, Matteoli M. Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J. 2020 Aug 17;39(16):e105380. Epub 2020 Jul 13 PubMed.
- Li T, Chiou B, Gilman CK, Luo R, Koshi T, Yu D, Oak HC, Giera S, Johnson-Venkatesh E, Muthukumar AK, Stevens B, Umemori H, Piao X. A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. EMBO J. 2020 Aug 17;39(16):e104136. Epub 2020 May 25 PubMed.
- Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014 Jun 6;344(6188):1164-8. PubMed.
- Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, Erreni M, Markicevic M, Starvaggi-Cucuzza C, Otero K, Piccio L, Cignarella F, Perrucci F, Tamborini M, Genua M, Rajendran L, Menna E, Vetrano S, Fahnestock M, Paolicelli RC, Matteoli M. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity. 2018 May 15;48(5):979-991.e8. Epub 2018 May 8 PubMed.
Further Reading
Primary Papers
- Das M, Mao W, Voskobiynyk Y, Necula D, Lew I, Petersen C, Zahn A, Yu GQ, Yu X, Smith N, Sayed FA, Gan L, Paz JT, Mucke L. Alzheimer risk-increasing TREM2 variant causes aberrant cortical synapse density and promotes network hyperexcitability in mouse models. Neurobiol Dis. 2023 Oct 1;186:106263. Epub 2023 Aug 15 PubMed.
- Rueda-Carrasco J, Sokolova D, Lee SE, Childs T, Jurčáková N, Crowley G, De Schepper S, Ge JZ, Lachica JI, Toomey CE, Freeman OJ, Hardy J, Barnes SJ, Lashley T, Stevens B, Chang S, Hong S. Microglia-synapse engulfment via PtdSer-TREM2 ameliorates neuronal hyperactivity in Alzheimer's disease models. EMBO J. 2023 Oct 4;42(19):e113246. Epub 2023 Aug 14 PubMed. Correction.
- Tzioras M, Daniels MJ, Davies C, Baxter P, King D, McKay S, Varga B, Popovic K, Hernandez M, Stevenson AJ, Barrington J, Drinkwater E, Borella J, Holloway RK, Tulloch J, Moss J, Latta C, Kandasamy J, Sokol D, Smith C, Miron VE, Káradóttir RT, Hardingham GE, Henstridge CM, Brennan PM, McColl BW, Spires-Jones TL. Human astrocytes and microglia show augmented ingestion of synapses in Alzheimer's disease via MFG-E8. Cell Rep Med. 2023 Sep 19;4(9):101175. Epub 2023 Aug 30 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.