Aβ Immunotherapy Revs Up Neurons in Mice—What About in People?
Quick Links
Hyperactive neurons that overheat brain circuits are par for the course in mouse models of AD and in people with the disease. A new study published in Nature Neuroscience on November 9 suggests that far from extinguishing these neuronal fires, antibodies against Aβ stoke them. The researchers, led by Arthur Konnerth at the Technical University of Munich in Germany, found that the hyperactivity occurs regardless of any reduction in amyloid burden or even the presence of plaques. The researchers have no explanation for the effect, although initial experiments make neuroinflammation unlikely. The researchers proposed that the phenomenon explains the absence of convincing cognitive benefits thus far in Aβ immunotherapy trials.
Although neurons ultimately wither away and die in the later stages of AD, they ramp up their activity in some regions of the brain in the earlier stages. Far from a good thing, neuronal hyperactivity can discombobulate neuronal circuitry. Many labs have found that calcium signals are out of whack in AD mouse models, while electrophysiological recordings indicate “silent seizures” caused by Aβ (see Sep 2007 news; Sep 2008 news; May 2012 news).
Functional magnetic resonance imaging (fMRI) has detected elevated hippocampal activity in people with preclinical AD, which correlated with cortical thinning and brain atrophy (see Dec 2011 news). Treatment with the anti-epileptic drug levetiracetam reduced this hyperactivity and improved cognition (see Mar 2015 news). Researchers have assumed that reducing levels of Aβ with therapeutic antibodies would also calm the amyloid-associated hyperactivity, and the idea is being tested in the clinic.
First author Marc Aurel Busche and colleagues used live two-photon calcium imaging, a technique only Konnerth’s and a few other labs have mastered, to measure neuronal activity in transgenic mice injected with Aβ antibodies. They treated 12- to 17-month-old plaque-ridden PDAPP mice for three months with weekly injections of 3D6, the mouse forerunner of bapineuzumab. While the treatment strongly relieved amyloid plaque burden, neuronal hyperactivity surpassed that seen in animals treated with a control antibody. The frequency of calcium bursts in the cortex doubled following treatment, to a level nearly fivefold higher than in wild-type mice. The proportion of hyperactive neurons more than tripled, and nearly half of the neurons fired in an unusual, synchronous fashion—a pattern that suggests epileptic activity, and that rarely occurred in control mice. Treatment of wild-type mice with 3D6 had no effect on neuronal activity. These results suggested that binding the antibody to Aβ facilitated the boost in neuronal activity in the AD mice.
Overactive neurons were also abundant in five-month-old PDAPP mice, despite their lack of amyloid plaques. Treatment of these younger mice with a single dose of 3D6 elevated this neuronal hyperactivity, suggesting that the antibody’s effects were not strictly related to the presence or clearance of plaques.
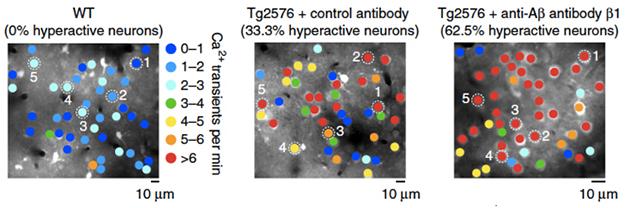
From Overdrive to Hyperdrive? In treated AD mice (right), hyperactive neurons (red) eclipse those in untreated (middle) and wild-type controls (left). [Courtesy of Busche et al., Nature Neuroscience 2015.]
To check their findings in another mouse model and another antibody, the researchers treated Tg2576 mice for three months with the antibody β1, which recognizes Aβ residues 3-6 and binds soluble, oligomeric, and fibrillar forms of the peptide, according to Busche. In keeping with previous reports that this antibody takes five months to budge plaques, the treatment left levels of soluble or insoluble Aβ in the brain unchanged. All the same, it substantially exacerbated neuronal hyperactivity (see image above). These results suggested that engagement of the antibody with Aβ, rather than the severity of amyloid pathology or the extent of plaque clearance, was key to elevating neuronal activity, said Busche.
What is the underlying mechanism? The researchers suspected neuroinflammation. However, a dose of the anti-inflammatory steroid dexamethasone failed to temper β1-mediated hyperactivity, while injections of the inflammation-stoking lipopolysaccharide did not make it worse. Antibody treatment did not raise levels of several proinflammatory cytokines in the cortex, as measured by ELISA. This suggested that β1 treatment did not substantially elevate neuroinflammation.
“The results of this study are surprising and thought-provoking,” said Donna Wilcock of the University of Kentucky. “They raise many more questions than they answer.” Wilcock felt more data was needed to rule out an effect of neuroinflammation. She said proinflammatory cytokines are notoriously difficult to detect in the brain, and the researchers used a standard ELISA kit that may have missed subtle increases (indeed, half of the cytokines the researchers measured, including IL-6, IL-10, and TNF-α, were undetectable in both groups of mice). She added that future studies could address one possible cause of neuroinflammation—the engagement of Fc receptors on glia and neurons. That could be tested by using antibodies lacking Fc regions or with their Fc regions altered to prevent activation, she said. Brian Bacskai of Massachusetts General Hospital in Charlestown agreed that the data could not rule out a role for neuroinflammation in the elevated neuronal hyperactivity.
If inflammation is not to blame, then how might treatment with Aβ antibodies ramp up neuronal hyperactivity? Busche speculated that the engagement of aggregated forms of Aβ by the antibodies could liberate soluble forms of Aβ, which could then stoke neuronal activity. Although the triggering of hyperactivity in young mice without plaques seems to contradict this idea, Busche said that even those mice likely harbor aggregated and/or fibrillar forms of Aβ that have not yet formed detectable plaques. Directly measuring such an effect in vivo would be difficult, he told Alzforum, as researchers would need to discern which forms of Aβ were released by the antibodies and which ones affected neurons. Busche said he plans to address this question indirectly by comparing multiple antibodies. While 3D6 and β1 bind both fibrillar and soluble forms of Aβ, other antibodies have a penchant for one versus the other (see May 2015 news). For example, solanezumab binds monomers, while aducanumab latches onto fibrils. Clinical trial results hint that both can slow cognitive decline in some patients (see Oct 2012 news; Aug 2015 conference news). Comparing hyperactivity in response to treatment with these and other antibodies could potentially test the liberation idea, and also point the way toward clinical antibodies that may be less likely to cause hyperactivity in trials.
“The study forces us to reconsider the importance of the epitope on the amyloid sequence to avoid or minimalize side effects,” commented Fred van Leuven of KU Leuven in Belgium. He added that 3D6 and β1 antibodies have been reported to bind full-length APP and C99 fragments, which could result in the binding and internalization of antibodies from the neuronal cell surface, perhaps causing calcium influxes.
Wilcock pointed out the elephant in the room—if Aβ immunotherapy is harmful, then why does it improve cognition in AD mouse models? The researchers did not perform behavioral experiments in this study. Busche said this was by design. “Mouse behavior may not translate into human behavior, and results from clinical trials indicate that mouse behavior is not useful for the evaluation of these treatments pre-clinically,” he said. He added that behavioral experiments often produce variable and controversial data.
Bacskai agreed that behavioral experiments can be hard to interpret and relate to human cognition. “On the other hand, one result they are clearly seeing is a bunch of neurons going crazy, and that’s got to amount to something,” he said. Busche added that unlike behavior, neuronal activity was a fundamental process that could more easily translate across species.
What about the claim that this neuronal overdrive explains, if even partly, the poor cognitive effects reported in most Aβ immunotherapy trials? Busche said that the hyperactivity could mask other benefits gleaned from Aβ reduction, at least until a substantial amount of Aβ was cleared. Clinicians could attempt to measure changes in synaptic activity using fMRI or EEG techniques in ongoing trials, Busche said.—Jessica Shugart
References
News Citations
- Do "Silent" Seizures Cause Network Dysfunction in AD?
- Hyperactive Neurons and Amyloid, Side by Side
- Soluble Aβ Takes Blame for Hyperactive Neurons in Mouse Brain
- Research Brief: Hippocampal Hyperactivity Tied to Early MCI Atrophy
- More Evidence That Epilepsy Drug Calms Neurons and Boosts Memory
- Shape of a Hug: How the Embrace of a Therapeutic Aβ Antibody Really Matters
- The Solanezumab Benefit: Oh, So Small, But Probably Real
- Aducanumab, Solanezumab, Gantenerumab Data Lift Crenezumab, As Well
Therapeutics Citations
Research Models Citations
Antibody Citations
Further Reading
Papers
- Busche MA, Konnerth A. Neuronal hyperactivity--A key defect in Alzheimer's disease?. Bioessays. 2015 Jun;37(6):624-32. Epub 2015 Mar 14 PubMed.
- Tischbirek C, Birkner A, Jia H, Sakmann B, Konnerth A. Deep two-photon brain imaging with a red-shifted fluorometric Ca2+ indicator. Proc Natl Acad Sci U S A. 2015 Sep 8;112(36):11377-82. Epub 2015 Aug 24 PubMed.
Primary Papers
- Busche MA, Grienberger C, Keskin AD, Song B, Neumann U, Staufenbiel M, Förstl H, Konnerth A. Decreased amyloid-β and increased neuronal hyperactivity by immunotherapy in Alzheimer's models. Nat Neurosci. 2015 Dec;18(12):1725-7. Epub 2015 Nov 9 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
KULeuven
This study examined the effects of chronic administration of two different anti-amyloid antibodies, 3D6 and β1, in two cohorts of two different mouse strains, PDAPP and Tg2576, which both stand as models for the amyloid component of Alzheimer’s disease. Although the study-design appears sub-optimal, the unexpected and interesting outcomes of both sets of experiments are (i) the increased neuronal activity induced by both antibodies 3D6 and b1, as measured by Ca2+imaging, and (ii) the lack of relation to amyloid burden, which was respectively decreased and unchanged by the two antibodies. These observations exclude also a potential gender effect, because the PDAPP cohorts contained only females and the Tg2576 were all male mice.
The surprise observation was that chronic treatment with either 3D6 or β1 triggered neuronal hyperactivity with higher frequency and in more neurons in both mutant APP mice. Higher synchronous firing in about half of the mice is taken by the authors to reflect the known incidence in AD patients of mild to severe epileptic symptoms. Seizure activity is evident in several amyloid mouse models and is held responsible for their high early death rate, and is, interestingly, strongly dependent on their genetic background.
There is no question that the outcome here is the opposite of what one expected, even more so because the exacerbated neuronal activity induced by both antibodies is unrelated to their effect on the amyloid burden in the three cohorts of old amyloid mice analyzed. While the observations are undisputable, much less so is the molecular mechanism inferred by the authors.
The pathology-related parameter measured as “amyloid burden,” equated to “amyloid plaques” in the text, was measured by immunohistochemistry (IHC) using the 3D6 antibody. This is actually the same as used for the treatment, which is somewhat remarkable. Biochemical data are provided only for soluble and insoluble Aβ40/42 in the Tg2576-β1 study, which are not affected. Biochemical data on various subtypes of amyloid peptides, from 38 to 42 and/or N-truncated or otherwise derivatized, and of the physical form known as oligomeric, would have been welcome. Their relevance eclipses that of “amyloid burden,” which is more and more depreciated as contributing essentially to the actual pathological process and cognitive demise in AD.
The highly scattered, variable levels of amyloid burden observed (Fig. 1a and Suppl. Fig. 2a) demonstrate this point even more since amyloid burden did not correlate with Ca2+ in the same mice (compare Fig 1a and b and Suppl Fig 2a and b). A scatterplot of both sets of data would have been illustrative and much appreciated.
Besides the molecular and physical diversity of amyloid peptides, we must consider intact APP as well as its major metabolites resulting from cleavage by BACE and ADAM10-C99 or beta-stubs, and C83 or alpha-stubs, respectively. Intact APP and C99 fragments are recognized by the 3D6 and β1 antibodies, which then would eventually decorate neurons and/or become internalized, contributing to the observed Ca2+ effects. Finally, one must also not forget the most recently reported metabolic cleavage of APP, called η-cleavage, as potentially contributing to the proposed mechanism involving cell-bound APP-metabolites.
Secondarily, the common induction of high neuronal activity by both antibodies was not observed in WT mice, nor was it related to inflammation, as measured by levels of pro-inflammatory cytokines in the brains of untreated and treated Tg2576 mice and in acute experiments using LPS and dexamethasone.
While the contribution of inflammation should not be considered as refuted by the data, the lack of human APP and all its metabolites named above in WT mice would explain the complete absence of any effects of these antibodies and supports the hypothesis that neuroinflammation was not involved.
Not least, the study offers advice or even warning about clinical applications of passive vaccination in AD patients, and forces us to (re)consider the importance of the selected epitope on the amyloid sequence to avoid or minimalize side effects. In this respect one is reminded that the Crenezumab antibody binds to the middle part of the amyloid peptide, inviting it to be tested in the same context as this study.
UK Dementia Research Institute@UCL and VIB@KuLeuven
I wonder whether the explanation that these antibodies bind partially with APP and APP CTF, which are heavily overexpressed in these mouse models, does not explain this finding. I would recommend expanding this study to include the knock-in mice from Takaomi Saido's group before making conclusions about what happens in people, in whom APP expression is much lower.
Make a Comment
To make a comment you must login or register.