Brain Injury Swells Ranks of Complement-Primed Microglia
Quick Links
Scientists have spotted a new variety of riled-up microglia that drives swelling in the human brain. In the November 18 Neuron, researchers from China led by Haixiao Liu, Shunnan Ge, and Yan Qu at Fourth Military Medical University in Xi’an, and Qiang Liu at Tianjin Medical University General Hospital, surveyed tissue from people undergoing surgery to ease pressure following a traumatic brain injury (TBI). Samples of swollen cortex teemed with inflammatory microglia expressing high levels of Complement C5a Receptor 1. In mouse models, deleting C5aR1 from microglia dialed down cytokine production and reduced fluid buildup, indicating that these complement-responsive cells are key instigators of the brain’s inflammatory response after a nasty knock.
- In people, inflammatory microglia surge after brain injuries.
- These cells express high levels of the complement receptor C5aR1.
- In a mouse model, knocking out microglial C5aR1 reduces inflammation and edema.
“One of the most striking aspects of this work is that the authors don’t just correlate a C5aR1-positive microglial state with severe brain injury—they show that genetically blocking C5a–C5aR1 dampens NF-κB-driven inflammatory programs …,” wrote Diane Chan, MIT, Cambridge, Massachusetts (comment below). “That makes the C5aR1 axis look much more like a causal driver of injury-associated pathology than a passive marker of ‘very sick’ tissue.”
Microglia greatly contribute to cerebral edema—the fluid swelling that follows acute brain injury. With single-cell sequencing, scientists are now beginning to sort out which specific microglial subtypes are involved and the molecular pathways they use.
Working out of Xi’an, first authors Jinpeng Zhou, Shuoyao Ma, Dayun Feng, and Yang Tian collected edema tissue from 16 people with traumatic brain injuries and from seven with brain bleeds during craniotomy, in which surgeons temporarily remove a piece of skull to ease pressure. For comparison, the researchers obtained six samples from individuals undergoing surgery to remove glioma tumors or abnormal clusters of blood vessels; to reach these lesions, surgeons remove a small piece of overlying healthy cortex that would otherwise be discarded. Of the 29 participants, 24 were male and five were female.
After dissociating cells from brain tissue, running RNA-Seq, and weeding out other cell types, the researchers recovered 96,113 microglial transcriptomes across all samples. Five distinct subtypes emerged based on the expression of known marker genes in both controls and brain-injury cases—homeostatic, inflammatory, stressed, ribosome-biogenesis, and phagocytic. In people with a TBI or brain bleed, the proportion of inflammatory microglia had expanded drastically compared with healthy cortex.
Relative to other microglia, this injury-related, inflammatory subtype upregulated 335 genes. Several encoded membrane receptors, among which C5aR1 was the most highly expressed. Compared with control cortex, microglia from injured tissue showed higher levels of C5aR1. This receptor binds C5a, a molecule released when the complement cascade is activated. C5a is a fragment of C5, which comes mostly from the liver and circulates in the blood. It sits downstream of proteins, including C1q and C3, that kick off the complement cascade. Together, these proteins recruit immune cells and help form the membrane attack complex, which punches holes in microbes.
To confirm these microglia made more of the receptor, Zhou probed TBI cortex with antibodies to C5aR1 and found it overlapped heavily with the microglial marker Iba-1. Far fewer C5aR1 positive microglia were present in control samples (image below).
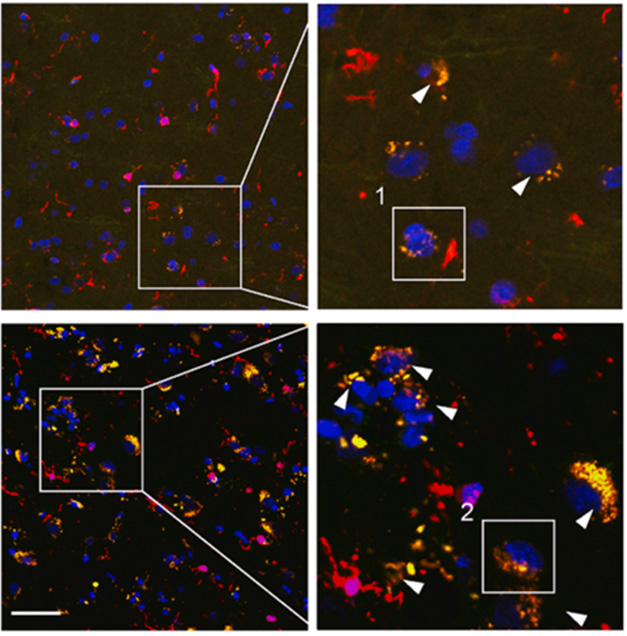
Traumatic Sign. C5aR1-positive microglia (orange) are more abundant in cortex from TBI tissue (bottom) than control (top). [Courtesy of Zhou et al., 2025, Neuron.]
Does C5aR1 stoke inflammation? To find out, the scientists switched the gene off in mouse microglia, then subjected the animals to a controlled brain injury. Three days later, the mice had less pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-6 in the brain than did C5aR1-expressing TBI controls. Magnetic resonance imaging revealed less edema (image below), suggesting that the complement-ready microglia are indeed important drivers of neuroinflammation in TBI.
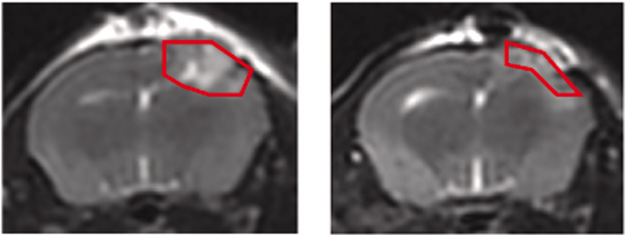
Sans C5aR1, Less Swelling. After a controlled brain injury, MRI shows more edema in control animals (left) than in C5aR1 knockouts (right). [Courtesy of Zhou et al., 2025, Neuron.]
It’s not yet clear whether C5aR1-expressing microglia play a similar role in other conditions, including neurodegenerative diseases such as Alzheimer’s, or even in ARIA—edema resulting from amyloid immunotherapy. Prior work hints they might.
Scientists led by Cynthia Lemere at Brigham and Women’s Hospital, Boston, reported that treating APP/PS1 mice expressing human APOE4 with amyloid-targeting antibodies activates the complement cascade along amyloid-laden blood vessels (Aug 2023 conference news; Mar 2024 conference news; Bathani et al., 2025). This may be a potential mechanism for ARIA, which occurs more often in people who carry the APOE4 allele. Without C3, fewer microglia became activated and cytokine production was toned down (Shi et al., 2017; Jun 2017 news). This suggests that, in mice, complement signaling is a normal part of the inflammatory microglial response to amyloid.
More recent work is consistent with this hypothesis. Scientists led by Anne Schaefer at the Max Planck Institute for Biology of Ageing in Cologne, along with Alexander Tarakhovsky at Rockefeller University and Alison Goate at the Icahn School of Medicine at Mount Sinai in New York City, found that in a mouse model of amyloidosis, expansion of a small subset of regulatory microglia prompted other microglia to dial down expression of complement-related genes (Ayata et al., 2025; Nov 2025 news).
This may be true in people, as well. “Looking at single-cell and single-nucleus studies of human Alzheimer’s cortex, we consistently see disease-associated microglial states that upregulate complement cascade genes, chemokines, and receptors, such as C5aR1, in proportion to amyloid and tau burden,” Chan told Alzforum.—George R. Heaton
George Heaton is a freelance writer in Durham, North Carolina.
References
News Citations
- Is ARIA an Inflammatory Reaction to Vascular Amyloid?
- Mouse Models and Markers for Cerebral Amyloid Angiopathy, ARIA
- Sans Complement: Amyloid Grows, Synapses and Memory Stay
- Protective Microglia Unmasked: They Act Like Regulatory T Cells
Paper Citations
- Bathini P, Schilling S, Lemere CA. Anti‐Amyloid Immunotherapy for AD: A Potential Link between ARIA and Complement. Alzheimer's and Dementia, January 3, 2025 Alzheimers & Dementia.
- Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, Lemere CA. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. 2017 May 31;9(392) PubMed.
- Ayata P, Crowley JM, Challman MF, Sahasrabuddhe V, Gratuze M, Werneburg S, Ribeiro D, Hays EC, Durán-Laforet V, Faust TE, Hwang P, Mendes Lopes F, Nikopoulou C, Buchholz S, Murphy RE, Mei T, Pimenova AA, Romero-Molina C, Garretti F, Patel TA, De Sanctis C, Ramirez Jimenez AV, Crow M, Weiss FD, Ulrich JD, Marcora E, Murray JW, Meissner F, Beyer A, Hasson D, Crary JF, Schafer DP, Holtzman DM, Goate AM, Tarakhovsky A, Schaefer A. Lymphoid gene expression supports neuroprotective microglia function. Nature. 2025 Dec;648(8092):157-165. Epub 2025 Nov 5 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Zhou J, Ma S, Feng D, Tian Y, Li L, Guo H, Shi Y, Cui W, Dong J, Hao S, Guo C, Xu M, Han L, Bai H, He D, Cui X, Chen L, Wu X, Gao F, Hu Q, Zhang Z, Wang K, Zhou G, Feng T, Xue D, Lin Q, Wang L, Gao L, Heng L, Zhao T, Luo T, Tian B, Zhang X, Guo W, Li Z, Liu H, Ge S, Liu Q, Qu Y. C5aR1+ microglia exacerbate neuroinflammation and cerebral edema in acute brain injury. Neuron. 2025 Nov 18; Epub 2025 Nov 18 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Alnylam Pharmaceuticals
To me, one of the most striking aspects of this work is that the authors don’t just correlate a C5aR1-positive microglial state with severe brain injury—they show that genetically or pharmacologically blocking C5a–C5aR1 signaling dampens NF-κB–driven inflammatory programs, reduces edema, and improves outcomes. That makes the C5aR1 axis look much more like a causal driver of injury-associated pathology than a passive marker of “very sick” tissue.
In terms of Alzheimer's disease, looking at single-cell and single-nucleus studies of human AD cortex, we consistently see disease-associated microglial states that upregulate complement cascade genes, chemokines, and receptors, such as C5aR1, in proportion to amyloid and tau burden. This is true in broader cortical atlases (for example, Mathys et al., 2019) as well as microglia-enriched datasets (Olah et al., 2020; Prater et al., 2023; Sun et al., 2023), which together define inflammatory and lipid-processing microglial states that look conceptually very similar to the edema-associated population described here. Given the prominent vascular injury, blood-brain barrier disruption, and microhemorrhage in ARIA and CAA, I would be surprised if a C5aR1-high inflammatory microglial phenotype were not also involved there, although we still lack direct human ARIA tissue to say this definitively.
With respect to 40 Hz stimulation, both our GENUS work, and others’ work show that gamma entrainment reshapes neuroimmune biology. In addition to microglial and astroglial changes in animal models, recent CSF proteomics after eight weeks of daily 40 Hz sensory stimulation in MCI demonstrated coordinated shifts in myelin-, synapse-, and neuroimmune-related proteins. Those data strongly suggest that gamma stimulation can modulate exactly the pathways that intersect with complement-driven microglial states, but the specific impact on C5aR1-positive microglia has not yet been mapped and is a very natural next step.
References:
Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai LH. Author Correction: Single-cell transcriptomic analysis of Alzheimer's disease. Nature. 2019 Jul;571(7763):E1. PubMed.
Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, Grün D, Kroshilina AA, Dionne D, Sarkis RA, Cosgrove GR, Helgager J, Golden JA, Pennell PB, Prinz M, Vonsattel JP, Teich AF, Schneider JA, Bennett DA, Regev A, Elyaman W, Bradshaw EM, De Jager PL. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer's disease. Nat Commun. 2020 Nov 30;11(1):6129. PubMed.
Prater KE, Green KJ, Mamde S, Sun W, Cochoit A, Smith CL, Chiou KL, Heath L, Rose SE, Wiley J, Keene CD, Kwon RY, Snyder-Mackler N, Blue EE, Logsdon B, Young JE, Shojaie A, Garden GA, Jayadev S. Human microglia show unique transcriptional changes in Alzheimer's disease. Nat Aging. 2023 Jul;3(7):894-907. Epub 2023 May 29 PubMed.
Sun N, Victor MB, Park YP, Xiong X, Scannail AN, Leary N, Prosper S, Viswanathan S, Luna X, Boix CA, James BT, Tanigawa Y, Galani K, Mathys H, Jiang X, Ng AP, Bennett DA, Tsai LH, Kellis M. Human microglial state dynamics in Alzheimer's disease progression. Cell. 2023 Sep 28;186(20):4386-4403.e29. PubMed.
Stanford University
This impressive study identifies a previously unrecognized C5aR1-positive microglial subtype that becomes markedly expanded in human cerebral edema tissue after TBI or intracerebral hemorrhage (ICH). Using single-cell transcriptomic profiling from human patients, the authors show that this subtype represents the dominant, pro-inflammatory microglial population in acute brain injury. Notably, the abundance of C5aR1-positive microglia correlates strongly with clinical severity, establishing it as a clinically relevant injury-associated microglial state.
Mechanistically, the study reveals that C5aR1-positive microglia amplify neuroinflammation by integrating C5a signals derived from both the injured brain and the liver, activating NF-κB, AKT, and ERK pathways to produce IL-1β, TNF-α, and chemokines such as CCL3 and CCL5. It is particularly interesting that both local and liver-derived C5a contribute to this process; however, the mechanism driving increased hepatic C5a production after brain injury remains to be determined.
C5aR1-positive microglia further promote neurotoxic A1 astrocyte polarization through classical microglial cytokines (TNF-α, IL-1α, C1q) as well as a newly implicated LGALS9–CD44 signaling pathway. In parallel, they orchestrate neutrophil recruitment into the injured brain via the CCL3-CCR1 and CCL5-CCR1 axes, forming a glia–immune communication loop that exacerbates cerebral edema.
Importantly, the study demonstrates that both genetic ablation of microglial C5ar1, and pharmacological inhibition using the FDA-approved C5aR1 antagonist avacopan, significantly reduce cerebral edema, preserve blood–brain barrier integrity, and improve neurological recovery in TBI and ICH mouse models. Together, these findings establish C5aR1-positive microglia as a central driver of neuroinflammation-induced cerebral edema and highlight microglial C5aR1 as a promising therapeutic target for acute brain injury.
University of California, Irvine
While some important details of the experiments are not clear, this paper presents a lot of approaches that, if performed and reported as assumed, demonstrate that at least some microglia that express C5aR1 contribute to neuroinflammation, pathology, and loss of function in two models of brain injury, TBI and ICH. A role for complement is very consistent with what has previously been published for TBI (Alawieh et al., 2021; Alawieh et al., 2018), but the advancement here is demonstrating that C5aR1 contributes substantially to the detrimental consequences and is quite consistent with what has been seen in preclinical models of Alzheimer’s disease (Schartz et al., 2024) and ALS (Lee et al., 2017; Ge et al., 2025). Altogether, the data provides more support for initiation of clinical trials with therapeutics targeting C5aR1 in numerous CNS disorders.
Some points from the manuscript:
References:
Alawieh A, Chalhoub RM, Mallah K, Langley EF, York M, Broome H, Couch C, Adkins D, Tomlinson S. Complement Drives Synaptic Degeneration and Progressive Cognitive Decline in the Chronic Phase after Traumatic Brain Injury. J Neurosci. 2021 Feb 24;41(8):1830-1843. Epub 2021 Jan 14 PubMed.
Alawieh A, Langley EF, Weber S, Adkins D, Tomlinson S. Identifying the Role of Complement in Triggering Neuroinflammation after Traumatic Brain Injury. J Neurosci. 2018 Mar 7;38(10):2519-2532. Epub 2018 Feb 6 PubMed.
Schartz ND, Liang HY, Carvalho K, Chu SH, Mendoza-Arvilla A, Petrisko TJ, Gomez-Arboledas A, Mortazavi A, Tenner AJ. C5aR1 antagonism suppresses inflammatory glial responses and alters cellular signaling in an Alzheimer's disease mouse model. Nat Commun. 2024 Aug 15;15(1):7028. PubMed.
Lee JD, Kumar V, Fung JN, Ruitenberg MJ, Noakes PG, Woodruff TM. Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. Br J Pharmacol. 2017 Apr;174(8):689-699. Epub 2017 Mar 3 PubMed.
Ge TQ, Wang P, Guan PP. Targeting the C5-C5aR1 axis: A promising therapeutic strategy for Alzheimer's disease and amyotrophic lateral sclerosis by unlocking neuroprotection. Biochem Pharmacol. 2026 Jan;243(Pt 1):117518. Epub 2025 Nov 2 PubMed.
Picower Institute of MIT
MIT Picower Institute for Learning and Memory
Microglial involvement in brain edema after acute injury is increasingly recognized as a major determinant of neurological outcome, but the field still lacks clarity on whether there are specific microglial populations that drive pathology because of their heterogenous nature. Zhou and colleagues contributed to a deeper understanding of microglial heterogeneity by identifying a novel C5aR1-expressing microglial subset that becomes highly enriched in human edema tissue after traumatic brain injury (TBI) on intracerebral hemorrhage (ICH). This highly pro-inflammatory microglial population activates astrocytes toward a neurotoxic phenotype and recruits neutrophils via CCL3/CCL5 signaling, driving a feed-forward cycle of blood-brain barrier breakdown and edema. Previously we identified CCL3-positive microglia (MG10) in early AD pathology (Sun et al., 2023). C5aR was also shown to be elevated in plaque-associated microglia (Ager et al., 2010). Therefore, similar complement-responsive inflammatory microglial states appear across both acute brain injury and early stages of neurodegeneration.
Interestingly, blockage of C5aR1 using avacopan improved recovery and behavioral outcomes from TBI by significantly reducing neuroinflammation in the brain. An additional report supports the idea that antagonizing C5aR1 signaling plays a critical role in reshaping detrimental microglial states and their interaction with neurotoxic astrocytes in Alzheimer’s disease (Schartz et al., 2024). These convergent findings make it plausible that a defined complement-responsive microglial subset represents a shared pathological axis across diverse CNS insults.
One additional angle worth highlighting is the potential link to astrocyte-mediated glymphathic regulation. Excess neuronal debris after acute brain injury can exceed microglial clearance capacity at the acute stage, and impaired glymphathic function could further increase debris burden and further worsen outcomes. This raises the possibility that complement-dependent microglial states could also impair clearance pathways relevant to ARIA and CAA, where glymphathic dysfunction is increasingly implicated. Given the well-established role of upstream complement cascade (C1q and C3) in response to amyloid, it would be interesting to investigate whether this microglial subset could offer therapeutic opportunities in other neurodegenerative diseases.
References:
Sun N, Victor MB, Park YP, Xiong X, Scannail AN, Leary N, Prosper S, Viswanathan S, Luna X, Boix CA, James BT, Tanigawa Y, Galani K, Mathys H, Jiang X, Ng AP, Bennett DA, Tsai LH, Kellis M. Human microglial state dynamics in Alzheimer's disease progression. Cell. 2023 Sep 28;186(20):4386-4403.e29. PubMed.
Ager RR, Fonseca MI, Chu SH, Sanderson SD, Taylor SM, Woodruff TM, Tenner AJ. Microglial C5aR (CD88) expression correlates with amyloid-beta deposition in murine models of Alzheimer's disease. J Neurochem. 2010 Apr 1;113(2):389-401. PubMed.
Schartz ND, Liang HY, Carvalho K, Chu SH, Mendoza-Arvilla A, Petrisko TJ, Gomez-Arboledas A, Mortazavi A, Tenner AJ. C5aR1 antagonism suppresses inflammatory glial responses and alters cellular signaling in an Alzheimer's disease mouse model. Nat Commun. 2024 Aug 15;15(1):7028. PubMed.
Make a Comment
To make a comment you must login or register.