Protective Microglia Unmasked: They Act Like Regulatory T Cells
Quick Links
Good managers set the tone for the whole team, encouraging positive behavior from everyone around them. In the November 5 Nature, scientists introduce a subtype of microglia that appears to do just that. The scientists were led by Anne Schaefer at the Max Planck Institute for Biology of Ageing, Cologne, Germany, with Alexander Tarakhovsky at the Rockefeller University and Alison Goate at Icahn School of Medicine at Mount Sinai, both in New York City. Though sparse, these “manager” cells clustered around amyloid plaques in mice and shaped the behavior of surrounding microglia. The effect? Harmful inflammation was kept at bay.
- In amyloidosis mice, a smattering of microglia express immunoregulatory T-cell receptors.
- Boosting this subset suppresses amyloid and tau pathology, preserves synapses, and lengthens survival.
- Suppressing them unleashes inflammatory responses.
- Similar microglia surround plaques in postmortem AD brain.
How? These cells made less of the master microglial regulator PU.1. This freed up expression of receptors typically found on regulatory T cells. These immunomodulatory receptors allowed PU.1-low cells to coordinate the overall microglia response to amyloidosis, keeping interferon and complement-activating pathways in check. Eliminating PU.1-low microglia from mice tripled the number of their inflammatory brethren and tripled plaque load. By contrast, amplifying PU.1-low microglia lightened plaque load and preserved synapses. In postmortem Alzheimer’s brain sections, similar immunomodulatory microglia surround plaques, implying the findings could apply to people.
Scientists found the data compelling. “This is an impressive and thought-provoking study that adds a genuinely new dimension to how we think about microglial heterogeneity in Alzheimer’s disease,” Giulia Albertini and Bart De Strooper at KU Leuven, Belgium, wrote to Alzforum. Jonathan Kipnis at Washington University in St. Louis called the findings exciting. “This work opens a new direction for therapeutically tuning microglia toward regulation rather than destruction, bringing us closer to manipulating these cells for brain repair,” he wrote (comments below).
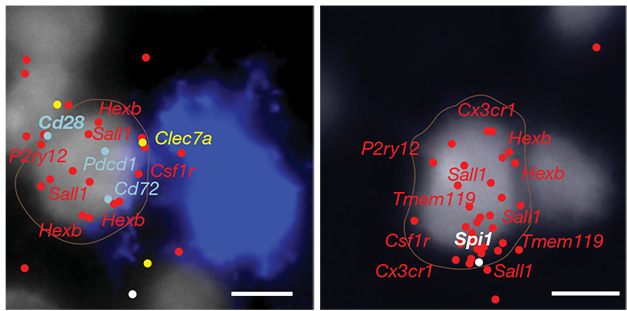
Plaque-Adjacent Signature. As seen via single-cell in situ hybridization, microglia (nuclei gray) near plaques (blue) express microglial genes (red), DAM genes (yellow), and lymphoid genes (light blue), but not SPI1 (white), the gene for the master regulator PU.1. Microglia far from plaques (right) express SPI1 but not lymphoid or DAM genes. [Courtesy of Ayata et al., 2025, Nature.]
Many previous studies have highlighted the dichotomous role of microglia in ameliorating or exacerbating amyloidosis and neurotoxicity. In mice, TREM2-expressing microglia help condense plaques and wall them off, protecting brain tissue (May 2016 news; Dec 2017 news; Sep 2019 news). Conversely, killing off microglia in 5xFAD mice, by blocking their survival factor CSF1, lowered inflammation, protected synapses, and preserved memory. The few microglia that remained in these mice were found next to plaques (Spangenberg et al., 2016; Mar 2018 news; Jul 2019 conference news). Schaefer, who was at Icahn until 2021, wondered whether these remaining microglia might be neuroprotective.
To investigate, joint first authors Pinar Ayata, Jessica Crowley, and Matthew Challman at Icahn isolated microglia from the cortices of 8-month-old 5xFAD mice and characterized them by single-cell RNA-Seq. Among the subset of disease-associated microglia (DAM), they found two distinct populations marked by either high or low expression of PU.1. Those with low PU.1 tended to cluster around plaques (image above). Notably, killing off microglia in 5xFAD mice using CSF1R inhibitors did not harm PU.1-low cells, suggesting this was indeed the subtype that had survived in prior microglial ablation experiments. Kim Green at the University of California, Irvine, who had done some of that work, agreed with this assessment (comment below).
What distinguished these nimble managers? Most notably, PU.1-low microglia expressed several immunomodulatory T-cell receptors such as CD28, PD1, CD5, and CTLA-2A (image below). They also boosted expression of lysosomal and lipid metabolism genes, suggesting they might have geared up to dispose of garbage. PU.1-low microglia were rare, making up less than 1 percent of the total.

To a “T.” A subtype of regulatory microglia express many of the immunomodulatory receptors typically found on T cells, such as CD28, PD1, CD5, and CTLA-2. Downstream, these receptors activate phospholipase Cγ. [Courtesy of Ayata et al., 2025, Nature.]
Because PU.1 is a core microglial determinant, Albertini and De Strooper wondered if PU.1-low cells can still be considered microglia. Schaefer believes they are, as the cells still express microglial genes. However, she thinks they undergo a “partial lineage relaxation” that allows them to express these T-cell receptors. Albertini and De Strooper also found it notable that previous single-cell RNA-Seq studies of microglia in 5xFAD mice had not identified these PU.1-low cells, probably because they are rare and mostly confined to plaques. “How many other microglial states are still sitting in plain sight but remain unrecognized because they do not conform to our usual clustering expectations?” they asked.
To find out what this small microglial subtype does, the authors knocked out one allele of SPI1, the gene encoding PU.1, in 5xFAD mice. This quadrupled CD28+ microglia and broadly calmed inflammation. Overall, microglia in these mice expressed half as much complement and interferon-response proteins as usual. They contained half as many lipid droplets as well.
“The selective regulation of the interferon-response state is intriguing and warrants further study,” noted Wei Cao at the University of Texas Health Science Center, Houston (comment below).
Their amyloidosis was less severe. By 10 months of age, these SPI1 heterozygotes had half as many plaques as 5xFAD controls. The plaques were compact, taking up two-thirds less area than typical. The PU.1-deficient 5xFAD mice maintained as many synapses as did wild-types, and they recognized new objects as readily. Notably, they lived four months longer than did regular 5xFADs.
“To our knowledge, this is the first study to show such a strong impact of microglia on the lifespan of mouse models of the disease,” Ayata, a co-corresponding author, told Alzforum.
Importantly, PU.1-low microglia seem to restrain tau pathology as well. When the authors injected aggregated human tau into the brains of 5xFAD mice lacking one SPI1 allele, the aggregates spread half as far as usual. Possibly, inflammatory microglia best propagate tau pathology, though this remains to be proven, Schaefer said.
By contrast, overexpressing PU.1 in microglia of 5xFAD mice shifted microglia toward inflammatory subtypes. Similarly, selectively deleting CD28 in microglia revved up expression of complement and interferon-response genes. Inflammatory microglia went from 10 to 30 percent of the total, and plaque load tripled. The authors did not report how this affected lifespan. To the authors, their data indicate that PU.1-low microglia broadly suppress the inflammatory response in other microglia. “We think they’re a regulatory cell population,” Schaefer told Alzforum. In future work, she will explore how this works.
Laís Ferreira and Soyon Hong at University College London noted that Schaefer’s data fit with a recent report that the immune-checkpoint receptor TIM-3 induces an anti-inflammatory state in microglia (May 2025 news). “[The data] suggest that known adaptive-immune checkpoints control a neuroprotective microglial state,” Ferreira and Hong wrote to Alzforum (comment below).
In that vein, Tarakhovsky noted that in the peripheral immune system, a small number of regulatory T cells control the inflammatory state of other immune cells. He thinks PU.1-low microglia serve the same function in the central immune system. “This discovery highlights shared principles of immune regulation,” he wrote to Alzforum.
How do PU.1-low microglia arise? Plaques themselves seemed to provoke this immune state by activating microglial surface receptors such as TREM2 and CLEC7A, the authors found. These receptors then switched on the tyrosine kinase SYK and its downstream target phospholipase Cγ2, a known protective factor. That, in turn, squelched PU.1 expression. In experiments, activating either TREM2, CLEC7A, or PLCγ2 suppressed PU.1, while knocking out SYK or PLCγ2 kept PU.1 high.
Schaefer believes these data may help to explain why having more TREM2, SYK, or PLCγ2 is neuroprotective against AD. “We think PU.1-low microglia are the executors of this protective signaling pathway,” she told Alzforum. Goate noted that the findings also offer a mechanistic reason for why a mutation that lowers PU.1 correlates with delayed AD onset (Huang et al., 2017).
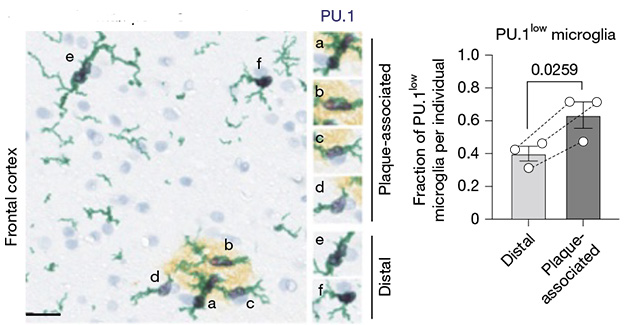
Dial It Down. In postmortem frontal cortex from an AD patient, microglia (green) adjacent (a, b, c, d) to plaques (gold) express less PU.1 (black) than do more distal microglia (e, f). Nuclei are blue. [Courtesy of Ayata et al., 2025, Nature.]
To start looking at whether this might happen in people, the authors analyzed existing RNA-Seq datasets from postmortem AD cortex (Dec 2020 news). They identified a subtype of microglia with lymphoid-like gene expression. Notably, this subtype increased in people carrying the protective PU.1-lowering mutation (Sep 2024 news). In addition, the authors immunostained postmortem frontal cortex sections from three AD patients and confirmed the presence of PU.1-low microglia around plaques (image above).
Schaefer and Tarakhovsky are interested in the therapeutic potential of these cells. Because such a small number of PU.1-low microglia are able to protect the brain, it might be possible to transplant enough cells into human brain to achieve a beneficial effect, Tarakhovsky suggested.
Michal Schwartz at the Weizmann Institute of Science in Rehovot, Israel, cautioned that complete loss of PU.1 has been found to be harmful. “The timing and the extent of PU.1 modulation seem critical,” she wrote. On that note, Nannan Lu and Tony Wyss-Coray at Stanford University, California, asked whether lowering PU.1 would still be beneficial late in AD. They also wondered how broadly these findings might hold. “[Is] the PU.1–CD28 axis specific to Alzheimer’s disease, or [does it] represent a general mechanism that regulates microglial function across development, aging, and other neurological disorders?” they asked (comments below).—Madolyn Bowman Rogers
References
News Citations
- Barrier Function: TREM2 Helps Microglia to Compact Amyloid Plaques
- Do Microglia Spread Aβ Plaques?
- Are Microglia Plaque Factories?
- Wiping Out Microglia Prevents Neuritic Plaques
- TREM2, Microglia Dampen Dangerous Liaisons Between Aβ and Tau
- Breaking the Spell: Checkpoint Inhibitors Help Microglia Snap Out of It
- Most Detailed Look Yet at Activation States of Human Microglia
- Glial Cell Communities Conspire to Drive Alzheimer’s Disease
Research Models Citations
Paper Citations
- Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, West BL, Green KN. Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid-β pathology. Brain. 2016 Apr;139(Pt 4):1265-81. Epub 2016 Feb 26 PubMed.
- Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, Harari O, Bertelsen S, Fairfax BP, Czajkowski J, Chouraki V, Grenier-Boley B, Bellenguez C, Deming Y, McKenzie A, Raj T, Renton AE, Budde J, Smith A, Fitzpatrick A, Bis JC, DeStefano A, Adams HH, Ikram MA, van der Lee S, Del-Aguila JL, Fernandez MV, Ibañez L, International Genomics of Alzheimer's Project, Alzheimer's Disease Neuroimaging Initiative, Sims R, Escott-Price V, Mayeux R, Haines JL, Farrer LA, Pericak-Vance MA, Lambert JC, van Duijn C, Launer L, Seshadri S, Williams J, Amouyel P, Schellenberg GD, Zhang B, Borecki I, Kauwe JS, Cruchaga C, Hao K, Goate AM. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer's disease. Nat Neurosci. 2017 Aug;20(8):1052-1061. Epub 2017 Jun 19 PubMed.
Further Reading
News
- New Evidence Confirms TREM2 Binds Aβ, Drives Protective Response
- TREM2: Diehard Microglial Supporter, Consequences Be DAMed
- Searching for New AD Risk Variants? Move Beyond GWAS
- The Mutation You Want: It Protects the Brain, Extends Life
- Janus-Faced PLCγ2? Alzheimer’s Risk Protein Toggles TREM2 and TLR Pathways
- PLCγ2 Variants Toggle Microglial Plaque Compactors
- SYK It To ’Em: Kinase Enables Microglia to Clear Plaques
- Plaque-Associated Microglia: A Villain’s Origin Story
- Glial Cell Communities Conspire to Drive Alzheimer’s Disease
Primary Papers
- Ayata P, Crowley JM, Challman MF, Sahasrabuddhe V, Gratuze M, Werneburg S, Ribeiro D, Hays EC, Durán-Laforet V, Faust TE, Hwang P, Mendes Lopes F, Nikopoulou C, Buchholz S, Murphy RE, Mei T, Pimenova AA, Romero-Molina C, Garretti F, Patel TA, De Sanctis C, Ramirez Jimenez AV, Crow M, Weiss FD, Ulrich JD, Marcora E, Murray JW, Meissner F, Beyer A, Hasson D, Crary JF, Schafer DP, Holtzman DM, Goate AM, Tarakhovsky A, Schaefer A. Lymphoid gene expression supports neuroprotective microglia function. Nature. 2025 Dec;648(8092):157-165. Epub 2025 Nov 5 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
UK Dementia Research Institute@UCL and VIB@KuLeuven
VIB/KU Leuven
This impressive and thought-provoking study adds a genuinely new dimension to how we think about microglial heterogeneity in Alzheimer’s disease. The identification of a PU.1low/CD28+ subset that is plaque-associated, TREM2-dependent, and resists CSF1R inhibition is a striking finding. It resonates with earlier observations from others of microglia that persist after treatment with PLX CSF1R inhibitors (for instance Baligács et al., 2024).
That said, we both found ourselves wanting more clarity on how this newly described population relates to those previously reported PLX-resistant cells. In our late-depletion PLX experiments, the remaining microglia do not appear to compensate and plaque burden can even increase, which complicates the interpretation. Are the PU.1low cells reported here the same population observed before, or does their behavior depend strongly on disease stage and context?
The lymphoid-like profile is particularly intriguing, as is the idea that PU.1 downregulation is induced by the plaque niche rather than being an intrinsic identity shift. Since PU.1 is such a core microglial determinant, this raises a bigger conceptual question: If a microglial cell can suppress PU.1 yet remain functional, and even protective, are we witnessing a deep reprogramming event or the emergence of a distinct cellular state?
We also found it notable that this “third axis” of DAM, with lymphoid-like characteristics, has not surfaced prominently in the many previous single-cell analyses of 5xFAD mice. It raises the possibility that analytical choices may have masked such states in earlier work. How many other microglial states are still sitting in plain sight but remain unrecognized because they do not conform to our usual clustering expectations?
Finally, although the authors report a reduction in plaque load, the mechanism remains a bit opaque. Based on the transcriptomic signature, these cells do not appear overtly phagocytic. Since tamoxifen induction occurs very early (4–6 weeks) and plaque assessments are done at 6 months, it is unclear whether the effect reflects active plaque removal or altered plaque dynamics over time (see also Baligács et al., 2024).
Overall, this is an excellent and stimulating paper. Beyond the specific findings, it rekindles the perennial question in our field: What truly makes a microglial response protective? Is it confinement of amyloid toxicity, preservation of broader microglial homeostatic functions, or something more systemic that we have yet to define?
References:
Baligács N, Albertini G, Borrie SC, Serneels L, Pridans C, Balusu S, De Strooper B. Homeostatic microglia initially seed and activated microglia later reshape amyloid plaques in Alzheimer's Disease. Nat Commun. 2024 Dec 5;15(1):10634. PubMed.
Washington University in St. Louis, School of Medicine
This is a really elegant, exciting, out-of-the-box study revealing that not all plaque-associated microglia are the same — some turn into suppressive, lymphoid-like cells that protect the brain. By lowering PU.1, Ayata and colleagues uncover a CD28-positive subset that calms inflammation, compacts plaques, and even limits tau spread — a remarkable demonstration that microglia can “re-educate” themselves into neuroprotective guardians. The genetic link is also appealing: an SPI1/PU.1-lowering allele in humans associates with more of these lymphoid gene–expressing microglia. This work opens a new direction for therapeutically tuning microglia toward regulation rather than destruction, bringing us closer to manipulating these cells for brain repair.
University of California, Irvine
This is a really nice and important study that will have broad implications for our understanding of the role of microglia and myeloid cells in AD pathogenesis. Microglial heterogeneity around plaques is interesting—it's clear that despite plaque reactivity, microglia (or at least myeloid cells) display different phenotypes and gene expression profiles despite being next to each other. Is this because they have different expression levels of PU.1, or maybe they originate from somewhere else (bone marrow)? Ten years ago we published that microglial depletion in 5xFAD mice via CSF1R inhibition depleted ~90 percent of microglia, but that a resistant population remained that was tightly associated with the plaque cores (Spangenberg et al., 2016). Even so, we saw overwhelmingly positive effects (rescue of synaptic loss, cognition, etc.). I always found it difficult to reconcile that we had depleted pretty much all the microglia except the ones reacting to plaques! This study now explains that the remaining microglia were these PU.1-low microglia that promote protection. This is a very satisfying result for me.
Mechanistically, there must be a reason why a subset of microglia around plaques express less PU.1 and transition to a protective phenotype. Identifying why this is may lead to new therapeutic avenues. Is it possible that these cells originated from the periphery and we could promote that somehow? Or is there an epigenetic mechanism at play that could be promoted? If so, maybe the cells transition to PU.1 high with time/disease progression?
References:
Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, West BL, Green KN. Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid-β pathology. Brain. 2016 Apr;139(Pt 4):1265-81. Epub 2016 Feb 26 PubMed.
University of Texas Health Science Center at Houston
This new study from Anne Schaefer’s group identifies a previously unrecognized microglial state enriched for lymphoid-associated genes that is neuroprotective in the context of amyloid pathology. Ayata et al. reach this conclusion through a tour de force that integrates in vitro and in vivo approaches—spanning genetic manipulation, lineage tracing, scRNA-Seq, spatial transcriptomics, CSF1R inhibition, tau seeding assays, and validation in human cell cultures and AD samples/datasets. Mechanistically, the work is particularly deep, outlining a cascade from plaque-engaged microglial surface receptors (TREM2/Clec7a/FcγR), through SYK/PLCG2 signaling, to PU.1 downregulation, induction of a lymphoid-like program, and CD28-mediated neuroprotection.
The genetic strategy used to probe PU.1 function is elegant and powerful. The authors engineered 5xFAD mice carrying one, two, or three copies of Spi1 (encoding PU.1) specifically in microglia, and reported complementary findings that support a gene-dose effect of PU.1 in shaping microglial states and downstream disease outcomes. These results not only confirm but also substantially extend prior genetic associations linking SPI1 variants (reduced PU.1 expression) to lower AD risk. As immune mechanisms continue to emerge as central in AD pathogenesis, it will be important to determine how PU.1 dosage influences the tauopathy phase of disease.
Two striking, cell-intrinsic effects of PU.1 dosing on microglial phenotypes emerge: lower PU.1 promotes formation of a microglial subset expressing lymphoid-related genes, whereas higher PU.1 favors accumulation of interferon-responsive microglia (IRM)—and these trends largely persist even without amyloid. We previously characterized IRM in 5xFAD (Roy et al., 2020; Roy et al., 2022); thus, the robust reduction of IRM in PU.1-low mice is especially notable. Because DAM clusters remain largely unchanged, the selective regulation of the IRM state is intriguing and warrants further study. Like PU.1, IRF8 is a core transcription factor (TF) in microglial development. Saeki et al. recently showed that IRF8-deficient microglia spontaneously upregulate IRM and DAM programs, underscoring the need for TFs in maintaining microglia homeostasis (Saeki et al., 2024). Given evidence that PU.1 and IRF8 interact and co-occupy ETS/ISRE enhancer regions in postnatal microglia (Kierdorf et al., 2013), a cooperative mechanism likely restrains spontaneous interferon signaling under baseline conditions.
The authors also identify CD28 as a marker of the PU.1-low, lymphoid-like microglial state, and demonstrate a functional role for this costimulatory receptor in suppressing inflammatory microglia, particularly IRM, and in mitigating AD progression. Notably, the CD28+ subset is relatively rare and appears later than DAM, IRM, and PU.1-low cells (after ~three months in 5xFAD). Yet microglial CD28 deletion precipitates excessive IRM formation even before overt microglial CD28 expression, an intriguing finding that invites further mechanistic dissection.
Overall, Ayata et al. delivered an exciting discovery with clear therapeutic implications. Analogous to immuno-oncology therapy strategies, the lymphoid-like program they describe highlights a set of overlapping immune-checkpoint targets with agents already approved or in clinical development. At the same time, careful evaluation will be needed to anticipate and minimize the adverse effects of immunotherapies when given to a large patient population (Vinnakota et al., 2024).
References:
Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, Propson NE, Xu Y, Jankowsky JL, Liu Z, Lee VM, Trojanowski JQ, Ginsberg SD, Butovsky O, Zheng H, Cao W. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. 2020 Apr 1;130(4):1912-1930. PubMed.
Roy ER, Chiu G, Li S, Propson NE, Kanchi R, Wang B, Coarfa C, Zheng H, Cao W. Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid β plaques. Immunity. 2022 May 10;55(5):879-894.e6. Epub 2022 Apr 19 PubMed.
Saeki K, Pan R, Lee E, Kurotaki D, Ozato K. IRF8 defines the epigenetic landscape in postnatal microglia, thereby directing their transcriptome programs. Nat Immunol. 2024 Oct;25(10):1928-1942. Epub 2024 Sep 23 PubMed.
Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Hölscher C, Müller DN, Luckow B, Brocker T, Debowski K, Fritz G, Opdenakker G, Diefenbach A, Biber K, Heikenwalder M, Geissmann F, Rosenbauer F, Prinz M. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013 Mar;16(3):273-80. Epub 2013 Jan 20 PubMed.
Vinnakota JM, Adams RC, Athanassopoulos D, Schmidt D, Biavasco F, Zähringer A, Erny D, Schwabenland M, Langenbach M, Wenger V, Salié H, Cook J, Mossad O, Andrieux G, Dersch R, Rauer S, Duquesne S, Monaco G, Wolf P, Blank T, Häne P, Greter M, Becher B, Henneke P, Pfeifer D, Blazar BR, Duyster J, Boerries M, Köhler N, Chhatbar CM, Bengsch B, Prinz M, Zeiser R. Anti-PD-1 cancer immunotherapy induces central nervous system immune-related adverse events by microglia activation. Sci Transl Med. 2024 Jun 12;16(751):eadj9672. Epub 2024 Jun 12 PubMed.
University College London
University College London
This is very elegant and beautiful work by Anne Schaefer, Pinar Ayata, and colleagues.
It is now increasingly clear that microglia have diverse functional states, especially in chronic neurodegenerative diseases such as Alzheimer’s. Even regarding plaques themselves, microglia have dual, if not more, roles, i.e., contributing to both amyloid plaque seeding and restricting disease progression (see for example Baligács et al., 2024). Hence, a key question is how to target the microglia that exacerbate disease while promoting disease-limiting states? However, the molecular programs of these microglial states and how they shift have been unclear.
Ayata and colleagues provide compelling evidence that microglia can shift toward neuroprotection in the plaque niche via regulation of PU.1, a transcriptional factor that defines microglia identity and function. A protective haplotype in the transcription factor PU.1 has previously been linked to delayed disease onset in Alzheimer’s disease (AD). Alison Goate and her team had shown that PU.1 knockdown dampened microglial inflammatory gene expression and phagocytosis (Huang et al., 2017). Here Ayata et al. show that PU.1low microglia are localized near amyloid plaques and adopt a transcriptional program enriched for lymphoid-associated genes, marked by CD28 expression. Microglia-specific PU.1 reduction attenuated cognitive decline, synapse loss, Aβ burden, and human tau propagation in 5xFAD mice. Conversely, microglia-specific CD28 deletion induced a type I interferon response, supporting the idea that PU.1 dosage tunes microglial immunosuppressive responses.
These data are exciting because they suggest we can potentially target specific microglia cell states. Overall, they support the view that microglia-targeted interventions could be beneficial and impactful in slowing neurodegeneration. Human validation studies will be the next important step.
The potential link to T cells and adaptive-innate crosstalk in neurodegeneration is also very exciting. A recent important study showed that depletion of immune-checkpoint receptor TIM-3 (HAVCR2), a T cell immune checkpoint controlling exhaustion, induces an anti-inflammatory state in microglia similar to PU.1 reduction (Kimura et al., 2025). An open question is whether CD28+PU.1low microglia are related to T cell interaction, recruitment and/or activation, but collectively, both studies endorse the relevance of investigating the plasticity of plaque-associated microglia, and they suggest that known adaptive-immune checkpoints control a neuroprotective microglial state.
Interestingly, Rustenhoven et al. previously showed that PU.1 knockdown reduces microglial antigen-presentation gene expression, indicating possible alterations in microglia-T cell communication (Rustenhoven et al., 2018). In addition to CD28, the PU.1low microglia also express PD-1 (Pdcd1), PD-L1 (Cd274), CD5, CTLA-2A, CD52, LAT2, and CD72. PD-1 is an immune checkpoint that controls adaptive immune activation and tolerance (Okazaki and Honjo, 2006). Notably, PD-1 blockade increases the number of T-regs restraining amyloid, and tau-mediated neurodegeneration, in mouse models (Baruch et al., 2016; Chen et al., 2023). It remains to be shown how inhibition of microglial-specific PD-1 and other lymphoid-related genes/checkpoints could affect microglia.
References:
Baligács N, Albertini G, Borrie SC, Serneels L, Pridans C, Balusu S, De Strooper B. Homeostatic microglia initially seed and activated microglia later reshape amyloid plaques in Alzheimer's Disease. Nat Commun. 2024 Dec 5;15(1):10634. PubMed.
Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, Harari O, Bertelsen S, Fairfax BP, Czajkowski J, Chouraki V, Grenier-Boley B, Bellenguez C, Deming Y, McKenzie A, Raj T, Renton AE, Budde J, Smith A, Fitzpatrick A, Bis JC, DeStefano A, Adams HH, Ikram MA, van der Lee S, Del-Aguila JL, Fernandez MV, Ibañez L, International Genomics of Alzheimer's Project, Alzheimer's Disease Neuroimaging Initiative, Sims R, Escott-Price V, Mayeux R, Haines JL, Farrer LA, Pericak-Vance MA, Lambert JC, van Duijn C, Launer L, Seshadri S, Williams J, Amouyel P, Schellenberg GD, Zhang B, Borecki I, Kauwe JS, Cruchaga C, Hao K, Goate AM. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer's disease. Nat Neurosci. 2017 Aug;20(8):1052-1061. Epub 2017 Jun 19 PubMed.
Kimura K, Subramanian A, Yin Z, Khalilnezhad A, Wu Y, He D, Dixon KO, Chitta UK, Ding X, Adhikari N, Guzchenko I, Zhang X, Tang R, Pertel T, Myers SA, Aastha A, Nomura M, Eskandari-Sedighi G, Singh V, Liu L, Lambden C, Kleemann KL, Gupta N, Barry JL, Durao A, Cheng Y, Silveira S, Zhang H, Suhail A, Delorey T, Rozenblatt-Rosen O, Freeman GJ, Selkoe DJ, Weiner HL, Blurton-Jones M, Cruchaga C, Regev A, Suvà ML, Butovsky O, Kuchroo VK. Immune checkpoint TIM-3 regulates microglia and Alzheimer's disease. Nature. 2025 May;641(8063):718-731. Epub 2025 Apr 9 PubMed.
Rustenhoven J, Smith AM, Smyth LC, Jansson D, Scotter EL, Swanson ME, Aalderink M, Coppieters N, Narayan P, Handley R, Overall C, Park TI, Schweder P, Heppner P, Curtis MA, Faull RL, Dragunow M. PU.1 regulates Alzheimer's disease-associated genes in primary human microglia. Mol Neurodegener. 2018 Aug 20;13(1):44. PubMed.
Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006 Apr;27(4):195-201. Epub 2006 Feb 24 PubMed.
Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, Kertser A, David E, Amit I, Schwartz M. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat Med. 2016 Feb;22(2):135-7. Epub 2016 Jan 18 PubMed.
Chen X, Firulyova M, Manis M, Herz J, Smirnov I, Aladyeva E, Wang C, Bao X, Finn MB, Hu H, Shchukina I, Kim MW, Yuede CM, Kipnis J, Artyomov MN, Ulrich JD, Holtzman DM. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023 Mar;615(7953):668-677. Epub 2023 Mar 8 PubMed. Correction.
Weizmann Institute of Science
This manuscript is particularly noteworthy, as it supports the growing interest in the use of immunotherapies for the treatment of Alzheimer’s disease. In this study, the authors focus specifically on microglia. While translating such approaches into viable immunotherapies remains challenging, especially in the context of microglia, the results presented here are scientifically compelling.
In brief, the authors focus on how microglial activity is regulated under AD conditions by PU.1, a major transcription factor of the myeloid lineage in general, and a determinant of microglial identity in particular. Somewhat paradoxically, lowering the levels of this master regulator appears to be protective in the context of AD. Yet, one should bear in mind that a complete loss of PU.1 expression is detrimental. Thus, both the timing and the extent of PU.1 modulation seem critical. The study also highlights the interplay between PU.1 activity and expression of inhibitory immune checkpoint pathways, such as PD-1/PD-L1/TIM-3, offering important insights into how fine-tuning microglial states could influence neurodegeneration.
Given the dynamics of microglia cells over the disease course, their heterogeneity across brain regions, and the fact that the same transcription factor regulates systemic myeloid cells, the findings are important, despite the challenges of clinical translation.
Stanford University Medical School
Stanford University
Microglia play a pivotal role in Alzheimer’s disease. Different subtypes of microglia have been shown to exert either protective or toxic effects during disease progression. However, the precise molecular mechanisms that determine whether microglia act in a helpful or harmful manner remain unclear.
This groundbreaking study from Anne Schaefer’s lab employs a broad array of complementary techniques, spanning mouse and human model systems, to reveal a previously unrecognized subpopulation of PU.1-low microglia. These microglia localize near amyloid plaques and are activated through a plaque-sensing pathway involving SYK and PLCγ2. Surprisingly, they upregulate multiple genes linked to immunoregulatory lymphoid receptor proteins, including CD28, thereby driving microglia into a neuroprotective state that suppresses neuroinflammation, limits amyloid plaque and tau accumulation, improves synapse density and synaptic plasticity, enhances cognitive performance, and extends lifespan in an amyloid mouse model. Studies with human brain tissue suggest the cells may exist in humans as well.
Earlier studies from Alison Goate’s lab identified common variants in SPI1, the gene encoding PU.1, as being associated with reduced Alzheimer’s risk. The work by Ayata et al. provides a detailed mechanistic explanation for how lower PU.1 levels might be linked to this protective effect.
Many exciting questions and ideas for future studies come immediately to mind. For example, molecules such as CD28, once thought to be exclusive to T and B lymphocytes, are minimally expressed in microglia under healthy conditions but become upregulated in AD, where they regulate microglial activity. The current findings highlight surprising parallels between suppressor T cells that prevent autoimmunity and PU.1-low, CD28-positive microglia that limit neuroinflammation in the brain. The main ligands for CD28 are CD80 and CD86, which are expressed on professional antigen-presenting cells including dendritic cells, macrophages, B cells and, in humans, on some activated T cells (and some microglia seem to be able to express them as well). Could such cells, possibly recruited to the inflammatory milieu of amyloid deposits, interact with and foster the emergence of CD28-expressing microglia?
More generally, what other convergent mechanisms are shared between the immune and nervous systems, and can we uncover and harness them for therapeutic applications in the near future, given that the cancer field has developed reagents against many of these immune modulatory factors?
Since the authors reduced PU.1 expression starting at 4–6 weeks of age (an early disease stage) in amyloid mouse models and observed beneficial effects, it will be very interesting to test whether reducing PU.1, enhancing the expression of CD28 and other lymphoid co-stimulatory, or co-inhibitory receptor proteins at mid- or late-disease stages could also produce therapeutic benefits.
A broader question is whether the PU.1–CD28 axis is specific to Alzheimer’s disease or represents a general mechanism that regulates microglial function across development, aging, and other neurological disorders.
In summary, this impressive work identifying the PU.1–CD28 axis establishes a molecular framework for understanding protective microglial states and sheds light on the potential of a new class of microglia-targeted immunotherapies to alter the course of Alzheimer’s disease.
Make a Comment
To make a comment you must login or register.