Does Loss of Neuronal Pentraxin 2 Start Network Dysfunction in Alzheimer’s?
Quick Links
What goes wrong first in the Alzheimer’s disease brain? Scientists led by Marc Aurel Busche of the U.K. Dementia Research Institute at University College London may have an answer. In the May 7 Neuron, they reported that before plaques formed in two different amyloidosis mouse models, neuronal activity in deep, but not higher, cortical layers went haywire. Excitatory neurons in layer 5/6 formed fewer synapses onto interneurons, dampening their inhibitory activity. This in turn unleashed excitatory neurons to fire more. The authors traced the root cause for all this to a shortage of the synaptic protein neuronal pentraxin 2 (NPTX2) in excitatory neurons. Overexpressing NPTX2 restored circuit activity to normal.
- Before plaques form in amyloidosis mice, excitatory neurons in layer 5/6 make less neuronal pentraxin 2.
- This weakens synaptic connections with inhibitory neurons, which become less active.
- Boosting NPTX2 restores normal network activity.
The data suggest that deep-layer neurons are particularly vulnerable to Aβ, Busche told Alzforum. “We see these changes even before we see hyperactivity in the superficial cortical layers,” he noted. He believes that restoring NPTX2 could be a promising strategy for early therapeutic interventions.
Inna Slutsky at Tel Aviv University, Israel, said the study expands scientists’ understanding of what goes wrong with brain circuitry early in AD. “The authors provide a compelling example of how early cortical inhibition failure can emerge in a layer- and cell-type-specific manner, revealing subtle but potentially pivotal circuit vulnerabilities that precede classical histopathological changes,” she wrote to Alzforum (comment below).

Rescuing Synapses. In layer 5/6 of wild-type mouse cortex (left), excitatory neurons form synapses (white box) on inhibitory neurons (gray circle), as seen by the overlay (yellow) of presynaptic marker VGLUT1 (red) and postsynaptic marker Homer1 (green). In 3-month-old APP/PS1 mice (middle), few synapses form. Overexpressing NPTX2 (right) restores them. [Courtesy of Papanikolaou et al., Neuron, 2025.]
Most previous investigations of brain circuitry have focused on superficial cortical layers, because these are easier to access. Studies by the authors and other groups have reported that layer 2/3 excitatory neurons near plaques become hyperactive in mouse models and AD brain (Sep 2008 news; Keskin et al., 2017; Gazestani et al., 2023). Scientists know less about what Aβ does to deeper cortical layers. These neurons have distinct properties, connecting to both cortical and subcortical structures, and firing more often than do upper neurons.
To explore what happens deep in the brain, first author Amalia Papanikolaou expressed a fluorescent calcium indicator in layer 5 mouse neurons and examined the cells by two-photon imaging. In 3-month-old APP/PS1 mice, excitatory neurons in the visual cortex fired off more bursts of action potentials, as seen by prolonged calcium transients, than did those in wild-type mice (image below). At the same time, parvalbumin-positive fast-spiking interneurons fired less often than normal. The same aberrant activity occurred in 3-month-old APPNL-G-F knock-in mice, which express endogenous levels of APP. Notably, at this age both mouse models had very little plaque, and layer 2/3 neurons maintained normal firing patterns.

Gone Wild. In 3-month-old wild-type mice (left), excitatory neurons in layer 5/6 have normal calcium transients (spikes). In APP/PS1 (middle) and APP-NL-G-F (right) mice, transients become abnormally prolonged (red spikes). [Courtesy of Papanikolaou et al., Neuron, 2025.]
What caused these activity changes? NPTX2 promotes excitatory synapses on interneurons by binding and stabilizing the AMPA receptor subunit GluA4 on these cells (Sia et al., 2007). Papanikolaou and colleagues measured the amount of both proteins in layer 5/6 of APP/PS1 mice via immunohistochemistry, and found less NPTX2 in excitatory neurons, and less GluA4 in interneurons than in wild-type mice. These changes correlated with the loss of two-thirds of excitatory synapses on interneurons. Overexpressing NPTX2 in excitatory neurons of the visual cortex with a viral vector doubled excitatory synapses on interneurons, boosted interneuron activity, and calmed excitatory neurons.
Could this happen in people? While there is no direct evidence from living brain, several studies have reported steep drops in NPTX2 in postmortem AD brain and in cerebrospinal fluid, with this loss correlating with cognitive decline (Xiao et al., 2017; Aug 2019 conference news; Apr 2025 news). Moreover, NPTX2 dwindles early, decades before memory problems show up in AD mutation carriers (Sep 2024 news).
Scientists had thought the NPTX2 scarcity reflected synapse loss in AD, but the new data suggest it precedes this loss, Busche noted. In ongoing work, he is exploring how Aβ suppresses NPTX2, and why this is layer-specific. He previously reported that oligodendrocytes release Aβ and contribute to neuronal hyperactivity (Oct 2024 news). Because oligodendrocytes become more abundant in deep cortical layers in AD, he thinks they might play a role. Busche will also investigate whether early circuit perturbations trigger later pathologies such as plaques and tangles.
Could selective vulnerability of deep cortical layers have implications for therapy? At the 2023 Alzheimer’s Association International Conference, Lars Lannfelt of Uppsala University, Sweden, reported that lecanemab preferentially binds to deeper cortical layers. In immunostainings of AD temporal cortex, most of the signal was confined to layers 4, 5, and 6. The data have not been published, and it is not clear why lecanemab has this specificity. “I’m still thinking about these data,” Busche said. “Do some of the differences between antibody efficacies depend on the layer specificity? It’s an interesting area for future investigation.” Nenad Bogdanovich, Karolinska Institute, Stockholm, who collaborates with Lannfelt, thinks one explanation could be that superficial layers may have more diffuse plaques, which contain less of the aggregated Aβ forms that lecanemab targets.
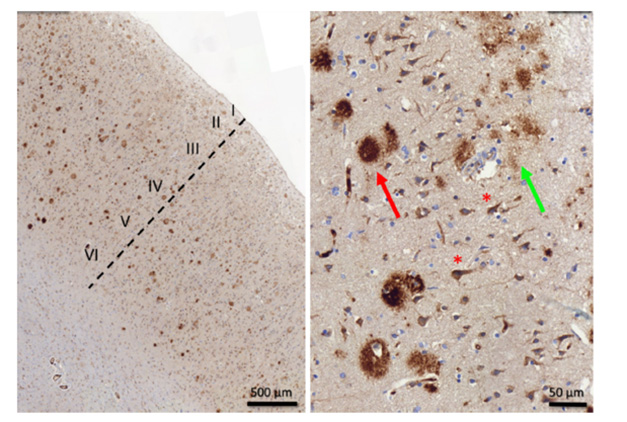
Going Deep. In immunostainings of postmortem AD temporal cortex, most lecanemab binds to deeper layers (left). In layer 3 (right), the antibody binds dense-core plaques (red arrow), diffuse plaques (green arrow), and intraneuronal Aβ (red asterisks). [Courtesy of Dan Sunnemark, Offspring Biosciences, and Lars Lannfelt.]
Slutsky noted that neuronal hyperexcitability in AD occurs more often during sleep than wakefulness. She would like to know whether the changes the authors found vary by brain region and physiological state. She believes the Neuropixels 1.0 probes Busche used could help address this question. These can record activity from almost 1,000 synapses for months. “Overall, this study provides a technically rigorous platform for probing the earliest stages of single-neuron and circuit-level dysfunctions in Alzheimer’s disease,” she wrote.—Madolyn Bowman Rogers
References
News Citations
- Hyperactive Neurons and Amyloid, Side by Side
- Synaptic Proteins in CSF: New Markers of Cognitive Decline?
- Do Synaptic Markers Foreshadow Cognitive Decline?
- CSF Proteins Spot Alzheimer’s Mutation Carriers Decades Before Symptoms
- Oligodendrocytes Pump Out Aβ42, Worsening Plaques and Synaptotoxicity
Research Models Citations
Therapeutics Citations
Paper Citations
- Keskin AD, Kekuš M, Adelsberger H, Neumann U, Shimshek DR, Song B, Zott B, Peng T, Förstl H, Staufenbiel M, Nelken I, Sakmann B, Konnerth A, Busche MA. BACE inhibition-dependent repair of Alzheimer's pathophysiology. Proc Natl Acad Sci U S A. 2017 Aug 8;114(32):8631-8636. Epub 2017 Jul 24 PubMed.
- Gazestani V, Kamath T, Nadaf NM, Dougalis A, Burris SJ, Rooney B, Junkkari A, Vanderburg C, Pelkonen A, Gomez-Budia M, Välimäki NN, Rauramaa T, Therrien M, Koivisto AM, Tegtmeyer M, Herukka SK, Abdulraouf A, Marsh SE, Hiltunen M, Nehme R, Malm T, Stevens B, Leinonen V, Macosko EZ. Early Alzheimer's disease pathology in human cortex involves transient cell states. Cell. 2023 Sep 28;186(20):4438-4453.e23. PubMed.
- Sia GM, Béïque JC, Rumbaugh G, Cho R, Worley PF, Huganir RL. Interaction of the N-terminal domain of the AMPA receptor GluR4 subunit with the neuronal pentraxin NP1 mediates GluR4 synaptic recruitment. Neuron. 2007 Jul 5;55(1):87-102. PubMed.
- Xiao MF, Xu D, Craig MT, Pelkey KA, Chien CC, Shi Y, Zhang J, Resnick S, Pletnikova O, Salmon D, Brewer J, Edland S, Wegiel J, Tycko B, Savonenko A, Reeves RH, Troncoso JC, McBain CJ, Galasko D, Worley PF. NPTX2 and cognitive dysfunction in Alzheimer's Disease. Elife. 2017 Mar 23;6 PubMed.
Further Reading
News
- Chicago: AD and Epilepsy—Joined at the Synapse?
- More Calcium News: Plaques Cause Dendrite Damage via Ion Overload
- Soluble Aβ Takes Blame for Hyperactive Neurons in Mouse Brain
- Cortical Biopsies Hint at Start of Alzheimer's 'Cellular Phase'
- Aβ Dimers Block Glutamate Uptake, Fire Up Synapses
- Proteomics Uncovers Potential Markers, Subtypes of Alzheimer’s
- Do Microglia Finish Off Stressed Neurons Before Their Time?
- CPTX—A Synaptic “Glue” Restores Memory in Alzheimer’s Model
- ‛iNET’ Cultures Expose NPTX2 as TDP-43 Henchman
- Neuronal Pentraxin 2 Binds Complement Protein, Protects Synapses
Primary Papers
- Papanikolaou A, Graykowski D, Lee BI, Yang M, Ellingford R, Zünkler J, Bond SA, Rowland JM, Rajani RM, Harris SS, Sharp DJ, Busche MA. Selectively vulnerable deep cortical layer 5/6 fast-spiking interneurons in Alzheimer's disease models in vivo. Neuron. 2025 Jul 23;113(14):2265-2279.e7. Epub 2025 May 8 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Tel Aviv University
This elegant study adds a crucial new dimension to our understanding of early circuit-level pathology in Alzheimer’s disease. By combining in vivo two-photon Ca²⁺ imaging with high-density single-unit recordings using Neuropixels probes in head-fixed mice, the authors identify a selective and early dysfunction of fast-spiking parvalbumin (PV) interneurons in cortical layer 5 (L5) of the V1 primary visual cortex, occurring prior to amyloid plaque deposition. Mechanistically, they demonstrate that PV hypoactivity is causally linked to a deep-layer-specific reduction of NPTX2 in excitatory neurons and GluA4 in PV interneurons, leading to a substantial loss of excitatory inputs onto these cells.
While previous work linked PV interneuron hypoactivity to network hyperexcitability and memory deficits (Palop and Mucke, 2016; Verret et al., 2012), this study stands out for its laminar precision and in vivo single-cell resolution, both rarely achieved in AD models. The authors provide a compelling example of how early cortical inhibition failure can emerge in a layer- and cell-type-specific manner, revealing subtle but potentially pivotal circuit vulnerabilities that precede classical histopathological changes.
As with any significant advance, the findings raise several thought-provoking questions:
Regarding the last question, several studies in the hippocampus (Kam et al., 2016; Slutsky, 2024; Soula et al., 2023; Zarhin et al., 2022) have shown that early network hyperexcitability in AD models is often state-dependent, emerging more prominently during sleep rather than wakefulness in early disease stages, and preceding cognitive symptoms. In contrast, Papanikolaou et al. report PV hypoactivity during wake states in V1 L5 of young AD model mice. Whether this discrepancy reflects fundamental differences in circuit function between the hippocampus and primary sensory cortices remains an open question.
The results also resonate with earlier studies of laminar-specific electrophysiology in V1 of healthy mice. For example, L5/6 excitatory neurons exhibit higher firing rates and lower burstiness in comparison to L2/3, and selective downregulation during NREM sleep, a feature not observed in superficial layers (Senzai et al., 2019).
It would be informative to know whether control mice in the Papanikolaou study display similar laminar dynamics, and how these properties are altered in the AD model, particularly during sleep, which was not examined here.
Future Neuropixels recordings across the sleep–wake cycle in freely behaving mice will be needed to determine whether early L5 PV interneuron dysfunction is a generalizable hallmark of AD pathology or varies with brain regions and vigilance states. Overall, this study provides a technically rigorous platform for probing the earliest stages of single-neuron and circuit-level dysfunctions in Alzheimer’s disease.
References:
Kam K, Duffy ÁM, Moretto J, LaFrancois JJ, Scharfman HE. Interictal spikes during sleep are an early defect in the Tg2576 mouse model of β-amyloid neuropathology. Sci Rep. 2016 Jan 28;6:20119. PubMed.
Palop JJ, Mucke L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci. 2016 Dec;17(12):777-792. Epub 2016 Nov 10 PubMed.
Senzai Y, Fernandez-Ruiz A, Buzsáki G. Layer-Specific Physiological Features and Interlaminar Interactions in the Primary Visual Cortex of the Mouse. Neuron. 2019 Feb 6;101(3):500-513.e5. Epub 2019 Jan 8 PubMed.
Slutsky I. Linking activity dyshomeostasis and sleep disturbances in Alzheimer disease. Nat Rev Neurosci. 2024 Apr;25(4):272-284. Epub 2024 Feb 19 PubMed.
Soula M, Maslarova A, Harvey RE, Valero M, Brandner S, Hamer H, Fernández-Ruiz A, Buzsáki G. Interictal epileptiform discharges affect memory in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A. 2023 Aug 22;120(34):e2302676120. Epub 2023 Aug 17 PubMed.
Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012 Apr 27;149(3):708-21. PubMed.
Zarhin D, Atsmon R, Ruggiero A, Baeloha H, Shoob S, Scharf O, Heim LR, Buchbinder N, Shinikamin O, Shapira I, Styr B, Braun G, Harel M, Sheinin A, Geva N, Sela Y, Saito T, Saido T, Geiger T, Nir Y, Ziv Y, Slutsky I. Disrupted neural correlates of anesthesia and sleep reveal early circuit dysfunctions in Alzheimer models. Cell Rep. 2022 Jan 18;38(3):110268. PubMed.
Vigil Neuroscience, Cambridge, Massachusetts
This is an exciting and timely study that adds an important layer to our understanding of early circuit dysfunction in Alzheimer’s disease. The finding that deep-layer (L5/6) parvalbumin-expressing interneurons exhibit suppressed spiking before overt plaque pathology, and that this is driven by reduced excitatory input resulting from decreased NPTX2 and GluA4 expression, highlights a spatially and cell-type-specific vulnerability that has not been fully appreciated.
What’s especially compelling is the demonstration that restoring NPTX2 expression in excitatory neurons not only rescues excitatory drive but also normalizes spiking of L5/6 PV interneurons. Interestingly, we showed previously that NPTX2 protects synapses by inhibiting complement activation and limiting microglia-mediated synapse pruning (Zhou et al., 2023). The convergence of molecular, physiological, and histopathological data across studies supports a model in which NPTX2 functions both as a coordinator of excitatory input to PV interneurons and as a safeguard against complement-mediated synaptic loss in AD.
The present study provides mechanistic insight linking NPTX2 loss to early inhibitory dysfunction and altered cortical information processing. This may help explain why reduced CSF NPTX2 levels are strongly associated with cognitive decline and predict the transition from asymptomatic to symptomatic AD (Soldan et al., 2023).
Looking ahead, it will be important to determine whether similar spatial- and cell-type-specific deficits occur in human AD, and whether therapeutic elevation of NPTX2 can help restore cortical inhibitory balance and preserve cognitive function.
References:
Zhou J, Wade SD, Graykowski D, Xiao MF, Zhao B, Giannini LA, Hanson JE, van Swieten JC, Sheng M, Worley PF, Dejanovic B. The neuronal pentraxin Nptx2 regulates complement activity and restrains microglia-mediated synapse loss in neurodegeneration. Sci Transl Med. 2023 Mar 29;15(689):eadf0141. PubMed.
Soldan A, Oh S, Ryu T, Pettigrew C, Zhu Y, Moghekar A, Xiao MF, Pontone GM, Albert M, Na CH, Worley P. NPTX2 in Cerebrospinal Fluid Predicts the Progression From Normal Cognition to Mild Cognitive Impairment. Ann Neurol. 2023 Oct;94(4):620-631. Epub 2023 Jul 25 PubMed.
University College London
This work by Papanikolaou et al. presents a series of neurophysiological investigations of neuronal activity across superficial and deep layers of primary visual cortex (V1) in AD mouse models.
The key findings from V1 deep layers (L5/6) of AD mouse models—prior to plaque deposition—include: increases in burst firing from excitatory neurons, which was associated with higher calcium load and metabolic cost; reductions of NPTX2 in excitatory neurons; and a ~70 percent decrease in excitatory synapse count on parvalbumin interneurons. The authors present evidence that NPTX2 overexpression mitigates layer-specific burst firing in APP/PS1 mice.
The findings suggest cellular mechanisms for underlying, and increasingly documented, posterior cortical abnormalities in AD—especially apparent in posterior cortical atrophy (“visual variant AD”). Of note:
While exercising caution regarding extrapolating neurophysiological findings from APP/PS1 mouse models to vulnerability of the human visual system in sporadic AD, the above prompts consideration of:
1) laminar-level, excitatory and interneuronal vulnerability in AD, perhaps owing to structural and functional characteristics, such as long-range projections and metabolic demands, and:
2) whether particular cellular vulnerability and/or recent evidence of a different microglial phenotype in posterior cortical atrophy (Abdi et al., 2025) relate to Nptx2 underexpression and microglia-mediated synapse loss.
References:
Hof PR, Bouras C, Constantinidis J, Morrison JH. Selective disconnection of specific visual association pathways in cases of Alzheimer's disease presenting with Balint's syndrome. J Neuropathol Exp Neurol. 1990 Mar;49(2):168-84. PubMed.
Tang-Wai DF, Graff-Radford NR, Boeve BF, Dickson DW, Parisi JE, Crook R, Caselli RJ, Knopman DS, Petersen RC. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology. 2004 Oct 12;63(7):1168-74. PubMed.
Abdi Z, Yong KX, Schott JM, Gatt A, Revesz T, Crutch SJ, Lashley T. Pathological Characterisation of Posterior Cortical Atrophy in Comparison With Amnestic Alzheimer's Disease. Neuropathol Appl Neurobiol. 2025 Apr;51(2):e70007. PubMed.
NeuDreamer LLC
This study represents a very important milestone, clarifying the intrinsic mechanisms related to sleep disturbances, the hyperexcitability of the hippocampus, and the excitotoxicity that leads to synapse and neuronal loss in the onset of Alzheimer's disease. Based on these mechanisms, the ability to selectively enhance the functional activity of parvalbumin (PV)-positive GABAergic inhibitory interneurons throughout the brain, including deep brain regions, has become a key to early AD treatment. Since the intrinsic oscillatory rhythm of PV-positive GABAergic inhibitory interneurons is the gamma rhythm (Hadler et al., 2024), deep brain-reachable, weak-intensity transcranial magnetic stimulation interventions targeting the function of PV-positive GABAergic interneurons may solve the problem (Zhen et al., 2017) and hold promise as an effective preventive and therapeutic approach for early AD.
References:
Hadler MD, Tzilivaki A, Schmitz D, Alle H, Geiger JR. Gamma oscillation plasticity is mediated via parvalbumin interneurons. Sci Adv. 2024 Feb 2;10(5):eadj7427. Epub 2024 Jan 31 PubMed.
Zhen J, Qian Y, Weng X, Su W, Zhang J, Cai L, Dong L, An H, Su R, Wang J, Zheng Y, Wang X. Gamma rhythm low field magnetic stimulation alleviates neuropathologic changes and rescues memory and cognitive impairments in a mouse model of Alzheimer's disease. Alzheimers Dement (N Y). 2017 Nov;3(4):487-497. Epub 2017 Sep 11 PubMed.
UEF
This paper investigates early circuit dysfunction in the visual cortex in a mouse model of Alzheimer's disease, focusing on the role of parvalbumin-positive (PV+) interneurons, the neuronal pentraxin 2 gene (NPTX2), and GluA4-containing AMPA receptors. The authors use a combination of large scale functional tools in vivo (two-photon calcium imaging and neuropixel single-cell electrophysiological probes), which enhanced single-cell resolution and provided structured laminar profiling of the cortical areas in an unprecedented manner compared to previous studies.
The authors report that in early stages of AD there is hypoactivity of PV+ interneurons in the deep, but not superficial layers of the visual cortex, accompanied by increased burst firing of putative pyramidal neurons. They identify a reduction in NPTX2 expression, which is crucial for clustering GluA4-containing AMPA receptors at excitatory synapses on PV+ interneurons. This reduction leads to impaired excitatory drive onto PV+ interneurons, disrupting the excitation-inhibition balance and contributing to network hyperexcitability and altered visual cortex processing (orientation/direction selectivity). Reinstating network functionality via adenoviral vector delivery of NPTX2 was able to restore the neurophysiological deficits in the visual microcircuit.
This paper strengthens the view that AD is a disorder of network dysfunction with synaptic integrity and input loss (rather than cell death) being the core change of early AD. It stresses that excitation inhibition balance, input coordination, and ultimately long-range subcortical connectivity (deep layer cortical output) are the downstream targets of such synaptic changes and emphasizes that not all synapses are equally vulnerable, pointing toward a synapse- and cell-type-specific degeneration model.
The reported dynamics of NPTX2 and GluA4 loss also testify toward a layer- and region-specificity (deep layers of V1 only, not L2/3) and reinforces the idea of microcircuit-specific vulnerability, not a general synaptic failure. Finally, this work stresses the predictive footprint that sensory cortices may have in early AD dysfunction and reframes the idea that the quantitative changes taking place in sensory processing areas can act as a readable output for early AD dysfunction and potential diagnosis biomarker.
This paper offers a beautifully detailed microcircuit model of early dysfunction, but whether this scales to the human brain remains unclear. The authors do a great job sketching out a molecular mechanism that leads to network failure, sensory dysfunction, and the proof-of-concept rescue of function. The manuscript raises a number of questions that are central to the quest for early AD biomarkers and calls for further validation of the links that it has provided, namely the generalizability of these ideas and the feasibility of a roadmap to early intervention in humans.
In particular, the authors do not explain why deep-layer interneurons in visual cortex are affected so early in this model. Is this a generalizable feature across cortical areas or specific to visual processing? Furthermore, they do not evaluate the extent of cross-species relevance of their findings nor whether the findings are related to this specific type of mouse model used. Our work finds that similar synaptic dysfunctionalities and hyperexcitable phenotypes occur more pronouncedly in the superficial rather than the deep layers of the human cortex (Gazestani et al., 2023; Dougalis et al., 2025). This raises the possibility of layer and cortical region-specific differences between mouse and human systems. If such laminar-specific discrepancies persist across models, it would suggest that current mouse paradigms may capture only aspects of the human pathology, warranting model diversification or adaptation.
The prodromal parieto-occipital atrophy subtype of human AD (Ten Kate et al., 2018) is associated with distinct biomarker changes. Unfortunately, Papanikolaou and colleagues do not provide biomarker analysis that could link the mouse model visual deficits (and the molecular mechanisms reported) to human pathology, so the translational relevance and the link to Aβ plaques or to later tau neurofibrils remain unknown. Additionally, the transient nature of the reported temporal dynamics of the NPTX2-GluA4 deficit (present only at around 3 months in the APP/PS1 mice but not at 1.5 or 6 months) implies that a critical window of vulnerability or plasticity failure exists. The proposed AAV-NPTX2 rescue, while conceptually compelling, faces translational barriers, not only due to the current limitations of viral delivery in humans (particularly for widespread cortical targeting), but also because of the narrow (and currently undefined in humans) time window of opportunity, which may be difficult to detect in otherwise asymptomatic clinical populations.
To facilitate interpretation of the findings by Papanikolaou and colleagues, and to better reveal the diagnostic and clinical value of their study, further cross-species and model validation of the proposed mechanisms are needed. This should include neuroimaging work on early dysfunction of sensory systems, combined with validation of the correlative and predictive value of biomarkers such as p-tau, Aβ, microglial markers, and NPTX2. Longitudinal blood and CSF sampling will also be critical in characterizing the temporal dynamics of the molecular changes described. Finally, evaluating whether CSF or plasma NPTX2 levels correlate with early functional, behavioral and structural abnormalities in sensory cortices (using tools such as EEG, MEG, or fMRI), particularly in the parieto-occipital regions implicated in prodromal AD, would strengthen the translational value of the study. There is a long road ahead, and this study has illuminated a promising path forward.
References:
Gazestani V, Kamath T, Nadaf NM, Dougalis A, Burris SJ, Rooney B, Junkkari A, Vanderburg C, Pelkonen A, Gomez-Budia M, Välimäki NN, Rauramaa T, Therrien M, Koivisto AM, Tegtmeyer M, Herukka SK, Abdulraouf A, Marsh SE, Hiltunen M, Nehme R, Malm T, Stevens B, Leinonen V, Macosko EZ. Early Alzheimer's disease pathology in human cortex involves transient cell states. Cell. 2023 Sep 28;186(20):4438-4453.e23. PubMed.
Dougalis A, Abushik P, Pelkonen A, Giudice L, Gomez-Budia M, Novosolova N, Valimaki N-N, Rezaie M, Nurkhametova D, Giniatullina R, Shakirzyanova A, Mali A, Rauramaa T, Stevens B, Hiltunen M, Leinonen V, Malm T. Early amyloid spine response and impaired synaptic transmission of pyramidal neurons in human biopsies with Alzheimer's Disease-related pathology. 2025 Jan 07 10.1101/2025.01.07.630516 (version 1) bioRxiv.
Ten Kate M, Dicks E, Visser PJ, van der Flier WM, Teunissen CE, Barkhof F, Scheltens P, Tijms BM, Alzheimer’s Disease Neuroimaging Initiative. Atrophy subtypes in prodromal Alzheimer's disease are associated with cognitive decline. Brain. 2018 Dec 1;141(12):3443-3456. PubMed.
Make a Comment
To make a comment you must login or register.