In Down’s Syndrome, Alzheimer’s Disease Has Unique Features
Quick Links
Is Alzheimer’s disease different in people with Down’s? Two recent proteomics studies say yes. In the July 1 Nature Communications, scientists led by Erik Johnson at Emory University School of Medicine, Atlanta, and Juan Fortea at the Biomedical Research Institute Sant Pau in Barcelona, Spain, reported that DSAD has a cerebrospinal fluid signature distinct from those signatures in autosomal-dominant and late-onset AD. Though Alzheimer’s biomarkers were comparable, neuroinflammation revved up earlier and faster in DSAD. Losses of myelin, inhibitory interneurons, and blood-brain barrier integrity also happened sooner, and were more pronounced.
- In DSAD, AD biomarkers and plaque proteins resemble those in other forms of AD.
- Abnormalities in the blood-brain barrier and extracellular matrix crop up earlier.
- Inhibitory interneurons fail more than a decade before symptom onset.
Complementing these findings, scientists led by Thomas Wisniewski at NYU Grossman School of Medicine, New York City, found that amyloid plaques from people with DSAD harbored a similar mix of proteins as did those from other forms of AD. However, brain tissue without plaque was strikingly different, with DSAD brains reflecting the same abnormalities that Johnson and Fortea saw in CSF. That work appeared in the January 18 Acta Neuropathologica. The two groups collaborated, with Wisniewski and Fortea listed as co-authors on the other’s papers.
“DSAD shows a lot of the same changes in proteomic markers as other types of AD, but in a unique sequence,” Johnson told Alzforum. That matters because it suggests people with DSAD might need different biomarkers and treatments. Wisniewski agreed. “There might be therapies that would be very specific for DSAD,” he speculated.
Other scientists said the data make a valuable contribution. “This comprehensive proteomic study provides novel insights into early pathological mechanisms in DS-associated AD,” Marc Busche and David Graykowski at University College London said of Johnson and Fortea’s study. Michael Rafii at the University of Southern California suggested the findings from both papers will facilitate clinical trials in DS and spur research. “This work will undoubtedly inspire more studies looking at proteomic changes at even earlier stages of the AD continuum in people with Down’s syndrome,” he wrote to Alzforum (comments below).

DSAD Over Time. In CSF from people with DS, some types of protein drop (blue) or rise (red) as much as 30 years before EYO (black line). [Courtesy of Montoliu-Gaya et al., Nature Communications, 2025.]
Leaky BBB, Crumbling Inhibitory Circuits Distinguish DSAD
Carrying three copies of the gene for amyloid precursor protein, people with DS inevitably develop Alzheimer’s pathology as they age. They accumulate amyloid plaques in their 30s and typically exhibiting symptoms around age 50 (Fortea et al., 2020). Previous studies found that DSAD closely matched other forms of AD. A spatial transcriptomics study of postmortem frontal cortex reported similar, but larger, changes in DSAD as in LOAD (Dec 2024 news). Meanwhile, the Alzheimer’s Biomarker Consortium-Down Syndrome found that a rise in plasma p-tau217 in people with DS portends encroaching cognitive decline, just as it does in other forms of AD (Jul 2022 news; Jun 2025 news). However, comprehensive proteomic studies of DSAD were lacking.
Johnson, Fortea, and colleagues took this on. Joint first authors Laia Montoliu-Gaya at the University of Gothenburg, Mölndal, Sweden, and Shijia Bian and Eric Dammer at Emory analyzed CSF samples from 229 participants in the Down Alzheimer Barcelona Neuroimaging Initiative. The cohort spanned the gamut from young people with no cognitive decline to those with full-blown dementia. Their mean age was 45. The authors compared these samples with those from 64 LOAD cases and 72 controls from the Sant Pau Initiative on Neurodegeneration cohort. The mean age of LOAD patients was 72, of controls, 57.
The authors ran tandem-mass-tag mass spectrometry on the samples, identifying a total of 838 proteins. In the people with DS, 556 were significantly higher or lower than in age-matched controls. Using age 50 as the estimated year of symptom onset (EYO), the authors placed DS changes on the time course of AD (image above). They found that classic AD biomarkers largely followed the traditional pattern, with Aβ42/40 dropping 15 years before EYO, p-tau217 and p-tau181 rising 10 years prior, and markers of astrocytosis and neuroinflammation going up five years prior.
Other proteins had distinct trajectories. In DSAD CSF, but not LOAD, ECM proteins, complement, and immunoglobulins shot up 20 years before EYO. Johnson told Alzforum these data could indicate a leaky blood-brain barrier that lets plasma proteins enter the brain. “Vascular changes appear quite early in DSAD,” Johnson said. Elizabeth Head at the University of California, Irvine, noted that this finding is consistent with other studies showing cerebrovascular disease developing with age in DS (Aug 2023 conference news; Lao et al., 2024).
Another early change? Thirteen years before EYO, parvalbumin, a marker of inhibitory interneurons, fell, while the neuronal injury marker NfL climbed. Loss of these neurons could explain why people with DS are prone to epilepsy, and might be a key feature of DSAD, Busche and Graykowski noted. “It provides a rationale for testing existing anti-epileptic medications, such as levetiracetam, in DS-associated AD,” they wrote.
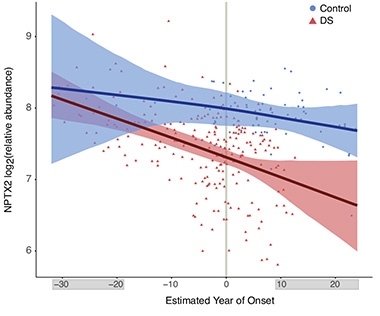
Early Synapse Loss? In CSF from people with DS (red), the synaptic marker NPTX2 falls below the levels in healthy controls (blue) 14 years before symptom onset. [Courtesy of Montoliu-Gaya et al., Nature Communications, 2025.]
One more surprise was that the synaptic marker NPTX2 dropped around the same time as changes in NfL and PVALB, whereas in LOAD, NPTX2 falls just before symptom onset (image at right). Vivek Swarup at the University of California, Irvine, said the early alterations in NfL, PVALB, and NPTX2 hint that damage to white matter and inhibitory circuitry could represent pivotal events in DSAD. “These CSF biomarkers are highly promising for staging disease and selecting candidates for early intervention,” Swarup wrote.
Johnson noted that the study was limited by the small size of the cohort, and by poor representation of younger people with DS, in particular. He plans to expand the cohort, adding more young people to determine which proteomic alterations predate DSAD. He also wants to switch from CSF to blood.
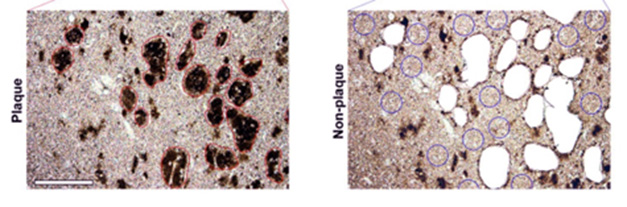
Precision Snips. In hippocampal sections from DSAD brain, scientists micro-dissected plaques (red circles, left) and nearby non-plaque tissue (blue circles, right) for proteomic analysis. [Courtesy of Martá-Ariza et al., Acta Neuropathologica, 2025.]
In DSAD Brain, the Extracellular Matrix is More Abnormal
Meanwhile, Wisniewski and colleagues directly examined brain tissue, building on their previous study in this area (Drummond et al., 2022). First author Mitchell Martá-Ariza used postmortem tissue from the hippocampus and the surrounding entorhinal and temporal cortex. Using a laser, he micro-dissected plaques and plaque-free tissue and identified the proteins in these samples by liquid chromatography-tandem mass spec (image above).
Wisniewski noted that the tissue came from several brain banks and was typically fixed with formalin and embedded in paraffin. Despite this, protein yields were as good as those from frozen tissue. “The method opens up tremendous stores of archival brain tissue for these sorts of investigations,” Wisniewski told Alzforum.
Martá-Ariza analyzed 20 samples each from DSAD, EOAD, LOAD, and control brains. The plaque proteome was consistent, with a correlation of about 0.75 between DSAD and the other two subtypes. The authors identified 43 plaque proteins that were common to all three diseases (image below). These were mainly proteins involved in APP metabolism, lysosomal functions, and immune response.

Imperfect Overlap. Plaques from DSAD, EOAD, and LOAD hippocampi had similar signatures, with 43 proteins consistently different in abundance compared with nearby plaque-free tissue (left). Plaque-free tissue varied more, with each disease showing distinct changes compared with control brain (right). [Courtesy of Martá-Ariza et al., Acta Neuropathologica, 2025.]
Nonetheless, there were surprises. The lysosomal ion channel CLCN6 was the most abundant protein in DSAD plaques, dwarfing the amount in EOAD and LOAD plaques. CLCN6 had not previously been identified as a plaque protein. Immunostaining showed this lysosomal protein building up in neurons at the edges of plaques. To the authors, this suggested it might participate in disgorging Aβ and building plaque. If so, it could be a therapeutic target, Wisniewski told Alzforum.
By contrast, non-plaque tissue varied greatly between diseases. DSAD stood apart from EOAD and LOAD, with a correlation of only 0.59 with the former and 0.33 with the latter (image above). In DSAD, ECM proteins were highly abundant, while in the other two diseases, DNA and RNA-binding proteins predominated. As in the CSF study, parvalbumin was down in DSAD. “We had fairly concordant findings overall,” Johnson noted of the two studies.
“These differences suggest that additional molecular pathways may drive earlier onset of AD or modify clinical trajectories in DS,” Head wrote. Marie-Claude Potier at the Institut du Cerveau–Paris Brain Institute said scientists need to put together a comprehensive picture of how AD varies across subtypes and biological compartments. “It is now essential to integrate data analyses from brain, CSF, and blood samples across various conditions, including DS, DSAD, LOAD, autosomal-dominant AD, and cerebral amyloid angiopathy, to further our knowledge and therapeutic strategies,” she wrote to Alzforum (comment below).—Madolyn Bowman Rogers
References
Mutations Citations
News Citations
- In Alzheimer’s Due to Down’s, Spatial Omics Spots Signs of Trouble
- In Down's Syndrome, Blood P-Tau217 Detects Plaques and Tangles
- Blood Tests Forecast Dementia in Down’s Syndrome
- At the Heart of Alzheimer’s in Down’s: Cerebrovascular Disease
Paper Citations
- Fortea J, Vilaplana E, Carmona-Iragui M, Benejam B, Videla L, Barroeta I, Fernández S, Altuna M, Pegueroles J, Montal V, Valldeneu S, Giménez S, González-Ortiz S, Muñoz L, Estellés T, Illán-Gala I, Belbin O, Camacho V, Wilson LR, Annus T, Osorio RS, Videla S, Lehmann S, Holland AJ, Alcolea D, Clarimón J, Zaman SH, Blesa R, Lleó A. Clinical and biomarker changes of Alzheimer's disease in adults with Down syndrome: a cross-sectional study. Lancet. 2020 Jun 27;395(10242):1988-1997. PubMed.
- Lao P, Edwards N, Flores-Aguilar L, Alshikho M, Rizvi B, Tudorascu D, Rosas HD, Yassa M, Christian BT, Mapstone M, Handen B, Zimmerman ME, Gutierrez J, Wilcock D, Head E, Brickman AM. Cerebrovascular disease emerges with age and Alzheimer's disease in adults with Down syndrome. Sci Rep. 2024 May 29;14(1):12334. PubMed.
- Drummond E, Kavanagh T, Pires G, Marta-Ariza M, Kanshin E, Nayak S, Faustin A, Berdah V, Ueberheide B, Wisniewski T. The amyloid plaque proteome in early onset Alzheimer's disease and Down syndrome. Acta Neuropathol Commun. 2022 Apr 13;10(1):53. PubMed.
Further Reading
Primary Papers
- Montoliu-Gaya L, Bian S, Dammer EB, Alcolea D, Sauer M, Martá-Ariza M, Ashton NJ, Belbin O, Fuchs J, Watson CM, Ping L, Duong DM, Nilsson J, Barroeta I, Lantero-Rodriguez J, Videla L, Benejam B, Roberts BR, Blennow K, Seyfried NT, Levey AI, Carmona-Iragui M, Gobom J, Lleó A, Wisniewski T, Zetterberg H, Fortea J, Johnson EC. Proteomic analysis of Down syndrome cerebrospinal fluid compared to late-onset and autosomal dominant Alzheimer´s disease. Nat Commun. 2025 Jul 1;16(1):6003. PubMed.
- Martá-Ariza M, Leitner DF, Kanshin E, Suazo J, Giusti Pedrosa A, Thierry M, Lee EB, Devinsky O, Drummond E, Fortea J, Lleó A, Ueberheide B, Wisniewski T. Comparison of the amyloid plaque proteome in Down syndrome, early-onset Alzheimer's disease, and late-onset Alzheimer's disease. Acta Neuropathol. 2025 Jan 18;149(1):9. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University College London
UCL UK Dementia Research Institute
This study provides a robust longitudinal analysis highlighting shared and distinct proteomic alterations, primarily in cerebrospinal fluid (CSF), from individuals with Down's syndrome and those with Alzheimer’s disease, complemented by comparative analyses of brain tissue.
While the proteomic analyses revealed many similarities, DS patients exhibited unique changes even before the onset of overt Aβ pathology. Of particular interest were modules associated with synaptic proteins, which declined notably earlier in DS compared to sporadic AD. One striking finding is the marked reduction of the activity-regulated protein Neuronal Pentraxin-2 (NPTX2). NPTX2 levels decreased significantly in both AD and DS cohorts, with a particularly pronounced and early reduction in DS. In line with this observation, our recent work identified early deep cortical layer-specific reductions of NPTX2 in APP mouse models, leading to reduced excitatory synaptic input specifically onto parvalbumin-positive (PV+) inhibitory interneurons, causing PV+ interneuron hypoactivity and a disrupted excitatory-inhibitory (E/I) balance (Papanikolaou et al., 2025). Alongside NPTX2, the authors also report reductions in other synaptic proteins (VGF and SCG2) known to regulate PV+ interneuron function (Selten et al., 2025), accompanied by a pronounced decrease in PV immunoreactivity itself.
Given the increased prevalence and risk of epilepsy in individuals with DS, combined with our group’s previous findings implicating neuronal hyperexcitability and altered E/I balance as key pathogenic mechanisms in AD (Harris et al., 2020), these observations point toward a potentially fundamental defect involving PV+ interneurons in DS-associated AD. The concurrent decreases in NPTX2, VGF, SCG2, and PV could thus represent key mechanistic hubs promoting neuronal network hyperexcitability and cognitive impairment in DS. Therapeutic strategies aimed at restoring the function of these interneurons and rebalancing E/I dynamics may therefore offer potential strategies for intervention in individuals with DS.
An additional finding is an early increase in immune and complement-related modules. Recent data indicate that NPTX2 also functions as a regulator of C1q, the initiating component of the classical complement cascade (Zhou et al., 2023). This raises the possibility of a mechanistic connection between early synaptic dysfunction and immune-mediated synaptic pruning. Future investigations should explore complement-mediated synapse elimination as a critical early event in DS-associated AD. Moreover, the authors highlight early cerebrovascular vulnerability in DS patients, notably through reduced MFGE8 levels, a protein strongly linked to cerebral amyloid angiopathy (CAA) and recently implicated in synapse elimination in APP mouse models (Sokolova et al., 2024). Given that cerebrovascular integrity is critical for maintaining stable neuronal networks, this raises questions about how early vascular dysfunction could interact with or exacerbate interneuron-related hyperexcitability. Clinically, the identified PV+ interneuron dysfunction and resulting E/I imbalance provide a rationale for testing existing anti-epileptic medications, such as levetiracetam, as targeted modulators of neuronal network function in DS-associated AD.
We contend that dysfunction of PV+ interneurons represents a central and early pathogenic event in DS-associated AD. The resulting disruption in E/I balance and consequent neuronal hyperexcitability could drive progressive dysfunction across neuronal networks, contributing to cognitive impairment, epilepsy, and neurodegeneration. Given similar observations in sporadic AD, PV+ interneuron dysfunction may reflect a common underlying mechanism and a broadly relevant therapeutic target.
In summary, this comprehensive proteomic study provides novel insights into early pathological mechanisms in DS-associated AD, notably emphasising PV+ interneuron dysfunction and complement-mediated synaptic loss as pivotal pathogenic processes. Further mechanistic studies addressing these hypotheses are eagerly anticipated.
References:
Harris SS, Wolf F, De Strooper B, Busche MA. Tipping the Scales: Peptide-Dependent Dysregulation of Neural Circuit Dynamics in Alzheimer's Disease. Neuron. 2020 Aug 5;107(3):417-435. Epub 2020 Jun 23 PubMed.
Papanikolaou A, Graykowski D, Lee BI, Yang M, Ellingford R, Zünkler J, Bond SA, Rowland JM, Rajani RM, Harris SS, Sharp DJ, Busche MA. Selectively vulnerable deep cortical layer 5/6 fast-spiking interneurons in Alzheimer's disease models in vivo. Neuron. 2025 Jul 23;113(14):2265-2279.e7. Epub 2025 May 8 PubMed.
Selten M, Bernard C, Mukherjee D, Hamid F, Hanusz-Godoy A, Oozeer F, Zimmer C, Marín O. Regulation of PV interneuron plasticity by neuropeptide-encoding genes. Nature. 2025 Jul;643(8070):173-181. Epub 2025 Apr 30 PubMed.
Sokolova D, Ghansah SA, Puletti F, Georgiades T, De Schepper S, Zheng Y, Crowley G, Wu L, Rueda-Carrasco J, Koutsiouroumpa A, Muckett P, Freeman OJ, Khakh BS, Hong S. Astrocyte-derived MFG-E8 facilitates microglial synapse elimination in Alzheimer's disease mouse models. bioRxiv. 2024 Dec 30; PubMed.
Zhou J, Wade SD, Graykowski D, Xiao MF, Zhao B, Giannini LA, Hanson JE, van Swieten JC, Sheng M, Worley PF, Dejanovic B. The neuronal pentraxin Nptx2 regulates complement activity and restrains microglia-mediated synapse loss in neurodegeneration. Sci Transl Med. 2023 Mar 29;15(689):eadf0141. PubMed.
University of Southern California Keck School of Mediicine
Just 10 years ago, it was still an open question whether Alzheimer’s disease in people with Down’s syndrome (DSAD) was fundamentally the same as in those with late-onset sporadic AD (LOAD) or autosomal dominant AD (ADAD). Thanks to the incredible progress made by large-scale efforts like DABNI and ABC-DS, we now have a much clearer picture. These rich datasets have shown us important similarities in the natural history and biomarker trajectories between DSAD and the other forms of AD and have allowed us to design and conduct clinical trials for DSAD.
These two recent papers using CSF proteomics take this understanding even further to provide a detailed comparison of their molecular aspects. Montoliu-Gaya and colleagues show that DSAD is marked by early immune activation, interneuron dysfunction, and cerebrovascular changes—long before amyloid plaques appear. The paper also highlights early declines in synaptic and myelin-related proteins, which may help explain the rapid cognitive decline and high prevalence of seizure activity, respectively, seen in DSAD.
Meanwhile, Martá-Ariza et al. reveal striking similarities in the plaque proteomes of DSAD, ADAD, and LOAD, while DSAD plaques are also highly enriched in proteins tied to the endo/lysosomal pathway. Specifically, the identification of CLCN6 as a prominent plaque-associated protein in DSAD opens up an exciting new therapeutic pathway.
The authors of both papers should be congratulated on this work, which will undoubtedly inspire more studies looking at proteomic changes at even earlier stages of the AD continuum in people with Down’s syndrome, the world’s largest genetically determined population with AD.
University of Kentucky
Research that includes people with DS helps us clarify the timing or sequence of events associated with AD initiation and progression. This very elegant study by Montoliu-Gaya et al. leveraged the DABNI cohort in a cross-sectional design and demonstrates that many proteomic changes occur in CSF prior to cognitive decline and prior to the gold standard AD biomarkers, e.g., Aβ. Concordance and discordance with brain tissue studies by Marta-Ariza et al., using similar approaches, was also valuable here.
It is striking how many of the proteomic CSF signatures in DS are similar to LOAD. This strongly suggests that this vulnerable and underserved community may benefit from similar treatments and access to clinical trials as the neurotypical population. The differences also are fascinating and not unexpected. Trisomy 21 involves many genes and pathways, including not only those related to AD, e.g. APP, but involved in oxidative stress (SOD1), mitochondrial dysfunction (ETS2), inflammation (IFNAR1/IL10), and white matter health (OLIG2), among others. Further, the difference in ECM proteins is consistent with reports of early and significant cerebrovascular disease with aging in people with DS (Lao et al., 2024). In DS, these differences also suggest that additional molecular pathways may drive earlier onset of AD, or modify clinical trajectories, and can serve as unique biomarkers for people with DS. The knowledge gained from this study not only informs AD in general but, importantly, helps us understand AD in people with DS.
The positive outlook from work such as this is that, given the strong age dependency in AD neuropathology but variability in the age of onset of clinical decline (with notable examples of resilience, see Liou et al., 2025), CSF biomarkers may provide novel, very early signals of potential decline or help predict individual trajectories. These results also suggest that a more targeted precision medicine approach, which for people with DS may be based upon the age of the individual in addition to CSF biomarkers, can target multiple pathways to potentially contribute to brain health.
Longitudinal studies using CSF proteomics as described here would expand our understanding of predictive value of the outcomes and how change in signatures may be consistent or different from LOAD and ADAD. For example, we know that AD progression in DS is more rapid, since there is a short interval between the accumulation of amyloid by PET and development of tau by PET (Wisch et al., 2024, and Zammit et al., 2024) and cognitive decline (Schworer et al., 2022). It will be exciting to relate the current observations from the DABNI cohort, but to use individual cognitive test scores to determine the delay from when proteomic signatures change and when different types/domains of cognition decline. Although necessary to compare ADAD and DSAD by using estimated year of onset, in DS there is also the opportunity to understand CSF proteomics changes with respect to age. Another future direction is to potentially use plasma in similar approaches since lumbar puncture, although entirely feasible, is more burdensome than a blood draw.
References:
Lao P, Edwards N, Flores-Aguilar L, Alshikho M, Rizvi B, Tudorascu D, Rosas HD, Yassa M, Christian BT, Mapstone M, Handen B, Zimmerman ME, Gutierrez J, Wilcock D, Head E, Brickman AM. Cerebrovascular disease emerges with age and Alzheimer's disease in adults with Down syndrome. Sci Rep. 2024 May 29;14(1):12334. PubMed.
Liou JJ, Lou J, Flores-Aguilar L, Nakagiri J, Yong W, Hom CL, Doran EW, Totoiu MO, Lott I, Mapstone M, Keator DB, Brickman AM, Wright ST, Nelson B, Lai F, Xicota L, Dang LT, Li J, Santini T, Mettenburg JM, Ikonomovic MD, Kofler J, Ibrahim T, Head E, Alzheimer Biomarker Consortium ‐ Down Syndrome. A neuropathology case report of a woman with Down syndrome who remained cognitively stable: Implications for resilience to neuropathology. Alzheimers Dement. 2025 Feb;21(2):e14479. Epub 2025 Jan 27 PubMed.
Wisch JK, McKay NS, Boerwinkle AH, Kennedy J, Flores S, Handen BL, Christian BT, Head E, Mapstone M, Rafii MS, O'Bryant SE, Price JC, Laymon CM, Krinsky-McHale SJ, Lai F, Rosas HD, Hartley SL, Zaman S, Lott IT, Tudorascu D, Zammit M, Brickman AM, Lee JH, Bird TD, Cohen A, Chrem P, Daniels A, Chhatwal JP, Cruchaga C, Ibanez L, Jucker M, Karch CM, Day GS, Lee JH, Levin J, Llibre-Guerra J, Li Y, Lopera F, Roh JH, Ringman JM, Supnet-Bell C, van Dyck CH, Xiong C, Wang G, Morris JC, McDade E, Bateman RJ, Benzinger TL, Gordon BA, Ances BM, Alzheimer's Biomarker Consortium-Down syndrome, Dominantly Inherited Alzheimer Network. Comparison of tau spread in people with Down syndrome versus autosomal-dominant Alzheimer's disease: a cross-sectional study. Lancet Neurol. 2024 May;23(5):500-510. PubMed.
Zammit MD, Betthauser TJ, McVea AK, Laymon CM, Tudorascu DL, Johnson SC, Hartley SL, Converse AK, Minhas DS, Zaman SH, Ances BM, Stone CK, Mathis CA, Cohen AD, Klunk WE, Handen BL, Christian BT, Alzheimer's Biomarker Consortium - Down Syndrome. Characterizing the emergence of amyloid and tau burden in Down syndrome. Alzheimers Dement. 2024 Jan;20(1):388-398. Epub 2023 Aug 29 PubMed.
Schworer EK, Zammit MD, Wang J, Handen BL, Betthauser T, Laymon CM, Tudorascu DL, Cohen AD, Zaman SH, Ances BM, Mapstone M, Head E, Christian BT, Hartley SL, Alzheimer's Biomarker Consortium–Down Syndrome (ABC–DS). Timeline to symptomatic Alzheimer's disease in people with Down syndrome as assessed by amyloid-PET and tau-PET: a longitudinal cohort study. Lancet Neurol. 2024 Dec;23(12):1214-1224. PubMed.
University of California, Irvine
The work by Montoliu-Gaya et al. demonstrates that DS-associated Alzheimer’s disease is driven by a complex, preclinical cascade—marked by immune activation, extracellular matrix remodeling, and blood–brain barrier perturbation—long before the onset of amyloid and tau pathology. Their finding that neurofilament light rises early, alongside a dramatic loss of interneuron and synaptic proteins, points to white matter injury and inhibitory circuit dysfunction as pivotal events in DSAD progression.
These CSF biomarkers are highly promising for staging disease and selecting candidates for early intervention; however, their translational value will hinge on longitudinal studies in larger, multiethnic DS cohorts, validation across peripheral fluids (e.g. plasma), and rigorous comparisons with sporadic and autosomal-dominant Alzheimer’s cases to establish sensitivity and specificity. Moreover, integrating these proteomic signatures with advanced neuroimaging and cognitive assessments will be essential to map molecular changes onto structural and functional decline, while mechanistic studies are needed to determine whether modulating immune–extracellular matrix interactions or bolstering interneuron resilience can truly alter the DSAD trajectory.
Paris Brain Institute
There are some notable findings in this study. The proteome of amyloid plaques is highly consistent across different diseases, though the proteome surrounding the plaques appears to differ between DSAD and both LOAD and EOAD. In the CSF, differences between DSAD and LOAD are also present.
A notable characteristic of DSAD, both around the plaques and in the CSF, is the increase in proteins encoded by chromosome 21 genes, extracellular matrix proteins, and myelin proteins. Parvalbumin, known as a marker for fast-spiking inhibitory interneurons, where APP transcripts are highly abundant (personal data), was enriched in amyloid plaques but showed reduced levels in the surrounding areas and in the CSF. The authors noted that there were no changes in parvalbumin levels in the brains of young individuals with DS, suggesting a potential link with AD in DS. This questions the role of APP or fragments in this specific interneuron subtype.
Recent reports indicate that APOE levels are elevated in the frontal cortex of individuals with DS (Farrell et al., 2025), likely due to the specific increase in the amyloid plaque proteome (Martá-Ariza et al., 2024). APOE levels were also elevated in CSF, raising questions about which proteins are inversely correlated between the amyloid plaque and the CSF proteomes, similar to the patterns observed with Aβ peptides.
The finding that most proteomic changes occur before the onset of AD symptoms, and concurrently with or before decreases in Aβ peptides and increases in total tau and p-tau217 in the CSF, is particularly promising for the discovery of earlier biomarkers in this population. This finding also highlights the challenges in correlating biomarkers in the CSF with changes in the brain, potentially due to dysfunctions in the blood-brain barrier and/or CSF barrier.
Additionally, comparing these results with the blood proteome will be crucial for a comprehensive understanding. It is now essential to integrate data analyses from brain, CSF, and blood samples across various conditions, including DS, DSAD, LOAD, ADAD, and CAA, to further our knowledge and therapeutic strategies.
References:
Farrell C, Buhidma Y, Mumford P, Heywood WE, Hällqvist J, Flores-Aguilar L, Andrews EJ, Rahimzadah N, Taso OS, Doran E, Swarup V, Head E, Lashley T, Mills K, Toomey CE, Wiseman FK. Apolipoprotein E abundance is elevated in the brains of individuals with Down syndrome-Alzheimer's disease. Acta Neuropathol. 2025 May 19;149(1):49. PubMed.
Martá-Ariza M, Leitner DF, Kanshin E, Suazo J, Giusti Pedrosa A, Thierry M, Lee EB, Devinsky O, Drummond E, Fortea J, Lleó A, Ueberheide B, Wisniewski T. Comparison of the amyloid plaque proteome in Down syndrome, early-onset Alzheimer's disease, and late-onset Alzheimer's disease. Acta Neuropathol. 2025 Jan 18;149(1):9. PubMed.
Icahn School of Medicine at Mount Sinai
My first thought is that Montoliu-Gaya and colleagues present a very interesting study of Down’s syndrome aging. Unlike autosomal-dominant Alzheimer’s disease, which has quite heterogeneous ages of onset due to the different severities of mutations, it seems that DS has a relatively consistent age of onset at 50, according to previous studies. So, in this study, estimated years until onset was set to the number of years until age 50. In other words, EYO and age are essentially equivalent here.
Thus, the early rise in inflammation and extracellular matrix proteins around age 30 (–20 EYO) in DS CSF suggests faster aging of these processes in DS. This is quite interesting, but whether these changes are truly linked to AD symptom onset is unclear. Future studies linking protein levels with actual age of AD symptom onset, adjusted for age, are needed to truly differentiate proteins related to DS aging versus Alzheimer’s disease.
Make a Comment
To make a comment you must login or register.