How Does a Neuron Avoid Aggregation of Liquid Protein Droplets?
Quick Links
Researchers believe that many proteins linked to neurodegenerative diseases function when condensed into liquid droplets. Unfortunately, for reasons that elude scientists, those droplets can harden into toxic protein aggregates. Two recent papers offer some insight into how a cell might regulate condensation and aggregation of proteins that undergo liquid-liquid phase transitions. Researchers led by Nicolas Fawzi at Brown University in Providence, Rhode Island, and Jeetain Mittal at Lehigh University, Bethlehem, Pennsylvania, drilled down to the atomic level to investigate. In the January 17 Molecular Cell, they reported that pathogenic mutations in the RNA-binding protein hnRNPA2 enhanced its propensity to form β-sheets, spurring fibrillization of droplets. On the other hand, methylation of hnRNPA2 disrupted protein contacts and slowed condensation.
- Pathogenic mutations in the ALS-linked protein hnRNPA2 promote β-sheet formation.
- Conversely, methylation of hnRNPA2 interferes with packing.
- Boosting autophagy can prevent mutant FUS from accumulating.
Meanwhile, researchers led by Jared Sterneckert at Technische Universität Dresden, Germany, found that revving up autophagy, a cellular disposal system, helped shrink RNA stress granules by degrading FUS, another protein that forms liquid droplets. Enhanced autophagy improved survival in cells and flies carrying mutant FUS, they wrote in the January 17 issue of Stem Cell Reports. Both hnRNPA2 and FUS have been linked to amyotrophic lateral sclerosis (ALS).
Fawzi believes these regulatory principles may apply to numerous neurodegenerative disease proteins, as they contain motifs and mutations similar to those found in hnRNPA2 and FUS. Others agreed, and said the findings point toward potential therapies. “That liquid-liquid phase transitions are regulated by posttranslational modifications is important, and suggests strategies for finding small molecules to restore normal RNA granule dynamics,” Paul Taylor at St. Jude Children’s Research Hospital in Memphis, Tennessee, wrote to Alzforum.
This research area exploded in 2015, when scientists discovered that RNA-binding proteins linked to neurodegenerative disease undergo liquid-liquid phase separation (Sep 2015 news; Oct 2015 webinar; Oct 2015 news). Researchers have identified 29 different RNA-binding proteins that contain the low-complexity domains (LCDs) that facilitate condensation, and many of these are involved in ALS, frontotemporal dementia, Alzheimer’s disease, and Huntington’s disease (King et al., 2012). Most recently, the list of molecules prone to liquid-liquid phase separation grew to include tau and even expanded-repeat RNAs (Oct 2015 conference news; Sep 2016 news; May 2017 conference news). However, the precise mechanism of condensation remains hazy.
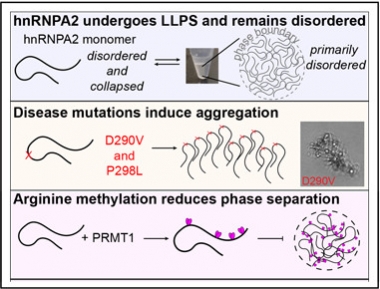
Droplet Dynamics.
Mutations and methylation affect the formation of liquid droplets and fibrillized aggregates. [Courtesy of Molecular Cell, Ryan et al.]
To shed some light, first author Veronica Ryan in Fawzi’s group used nuclear magnetic resonance spectroscopy to study hnRNPA2 interactions in cell-free solutions. She found that the hnRNPA2 LCD had little secondary structure and formed only fleeting connections with the low-complexity domains of other hnRNPA2 molecules. Salt forced hnRNPA2 LCDs to condense into droplets, likely held together by hydrophobic bonds. However, the LCDs remained disordered, perhaps explaining how the droplets remain fluid.
This changed when hnRNPA2 contained a pathogenic variant. Ryan tested the D290V mutation that associates with multisystem proteinopathy, an ALS-like disease. D290V replaces a charged aspartic acid in the low-complexity domain with a nonpolar valine. Liquid droplets of D290V hnRNPA2 quickly converted to fibrous aggregates. Computational analysis predicted that this mutation would cause the low-complexity domains to spoon each other, forming a β-sheet, in agreement with a previous study (Thompson et al., 2006; Shorter and Taylor, 2013).
By contrast, the P298L mutation associated with Paget’s disease of bone replaces a hydrophobic proline with another hydrophobic residue. Nevertheless, droplets of this variant also fibrillized, though more slowly than the D290V variant. How might this substitution promote aggregation? Fawzi noted that proline prevents β-sheet formation because it causes a kink in the protein backbone that interferes with packing. In addition, proline cannot form a hydrogen bond along the backbone, another key ingredient for β-sheet formation. The proline residue at this location is highly conserved, suggesting it is crucial for keeping protein gels liquid, Fawzi added.
What else might prevent packing? The authors examined methylation because it has been shown to affect phase separation of other proteins (Nott et al., 2015). In solution, the enzyme protein arginine methyl transferase 1 modified four arginine residues in hnRNPA2’s low-complexity domain. Methylated hnRNPA2 remained soluble at 50 percent higher protein concentrations than did wild-type hnRNPA2, indicating it is less prone to form droplets. Computer simulations led by Mittal’s group revealed that methylation disrupted contacts between the arginines and aromatic residues in the low-complexity domain, helping keep hnRNPA2 molecules separated.
Fawzi will next ask if these same interactions occur in cells, how they affect cellular function, and whether arginine methylation controls condensation of other RNA-binding proteins such as FUS and TDP-43.
Dorothee Dormann at Ludwig-Maximilians-Universität, Munich, found this intriguing. “Arginine methylation seems to be a particularly interesting modification, since it is often mis-regulated in disease, and defects have been identified in FUS-associated neurodegeneration,” she wrote to Alzforum. Arginine methylation could play a role in preventing liquid-liquid phase separation and aggregation in general, Dormann suggested (Sep 2012 conference news; Thandapani et al., 2015; Suárez-Calvet et al., 2016).
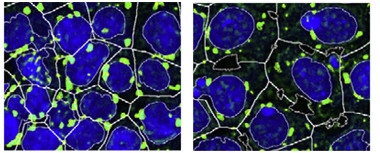
Granule Cleanup.
Induced pluripotent stem cells (nuclei blue) treated with an autophagy stimulator (right) accumulate fewer and smaller stress granules (green) during oxidation than do untreated cultures (left). [Courtesy of Stem Cell Reports, Marrone et al.]
Sterneckert and colleagues took a different approach to combating aggregation. They investigated the FUS P525L mutation, which disrupts FUS’ nuclear localization signal such that excess FUS accumulates in cytoplasm. When first author Lara Marrone inserted the mutation into the genomes of human induced pluripotent stem cells, it led to a fourfold increase in cytoplasmic FUS. These stem cells developed bigger and more numerous stress granules in response to oxidative or heat stress.
Then the authors screened a library of about 1,000 small molecules to find those that suppressed granule formation in the iPSCs after oxidative stress. Sixty-nine compounds shrunk the total stress granule area; 13 of these were inhibitors of the mTOR signaling pathway that suppresses autophagy. Investigating five of the compounds further, the authors found a dose-dependent protection against stress granules comparable to that of the mTOR inhibitor rapamycin (see image above). The compounds seemed to affect stress granules indirectly, because there was no sign of increased autophagy of these structures. Instead, the compounds may stimulate degradation of stray cytoplasmic FUS before it incorporates into stress granules, the authors suggested. In keeping with this, the compounds only worked when administered before granules were there.
Since mTOR inhibitors suppress immune function, Marrone looked for safer ways to turn up autophagy in the brain. She screened 1,600 approved drugs in the same assay. Of 70 that inhibited stress granule formation, several, mostly antipsychotics and antidepressants, were brain-penetrant. Six of these—paroxetine, promethazine, trimipramine, penfluridol, imipramine, and chlorpromazine—had been reported previously to boost autophagy and share a similar underlying structure (Tsvetkov et al., 2010). These drugs could be a starting point to develop more effective therapies, the authors suggested.
Sterneckert is now evaluating whether these drugs lower deposits in other ALS subtypes caused by mutations in TDP-43 and C9ORF72. Other consider this a promising direction, as many autophagy genes have been linked to ALS or other neurodegenerative diseases. “Drugs that promote the degradation of misfolded proteins, and thus prevent the conversion of stress granules into aggregates, hold great promise for the development of therapeutic approaches for several types of age-related neurodegenerative diseases,” Serena Carra at the University of Modena and Reggio Emilia, Italy, wrote to Alzforum. Sterneckert is hunting for a blood-based biomarker of autophagy.—Madolyn Bowman Rogers
References
News Citations
- ALS Protein Said to Liquefy, Then Freeze en Route to Disease
- Do Membraneless Organelles Host Fibril Nucleation?
- Does C9ORF72 Repeat RNA Promote Protein Phase Transitions?
- Helical Tail Holds Sway Over TDP-43 Packaging
- Protein Liquid-Liquid Phase Transitions: The Science Is About to Gel
- Arginine Methylation Distinguishes ALS-FUS From FTLD-FUS
Webinar Citations
Paper Citations
- King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012 Jun 26;1462:61-80. Epub 2012 Jan 21 PubMed.
- Thompson MJ, Sievers SA, Karanicolas J, Ivanova MI, Baker D, Eisenberg D. The 3D profile method for identifying fibril-forming segments of proteins. Proc Natl Acad Sci U S A. 2006 Mar 14;103(11):4074-8. PubMed.
- Shorter J, Taylor JP. Disease mutations in the prion-like domains of hnRNPA1 and hnRNPA2/B1 introduce potent steric zippers that drive excess RNP granule assembly. Rare Dis. 2013;1:e25200. Epub 2013 May 29 PubMed.
- Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD, Baldwin AJ. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell. 2015 Mar 5;57(5):936-47. PubMed.
- Thandapani P, Song J, Gandin V, Cai Y, Rouleau SG, Garant JM, Boisvert FM, Yu Z, Perreault JP, Topisirovic I, Richard S. Aven recognition of RNA G-quadruplexes regulates translation of the mixed lineage leukemia protooncogenes. Elife. 2015 Aug 12;4 PubMed.
- Suárez-Calvet M, Neumann M, Arzberger T, Abou-Ajram C, Funk E, Hartmann H, Edbauer D, Kremmer E, Göbl C, Resch M, Bourgeois B, Madl T, Reber S, Jutzi D, Ruepp MD, Mackenzie IR, Ansorge O, Dormann D, Haass C. Monomethylated and unmethylated FUS exhibit increased binding to Transportin and distinguish FTLD-FUS from ALS-FUS. Acta Neuropathol. 2016 Apr;131(4):587-604. Epub 2016 Feb 19 PubMed.
- Tsvetkov AS, Miller J, Arrasate M, Wong JS, Pleiss MA, Finkbeiner S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010 Sep 28;107(39):16982-7. PubMed.
Further Reading
Primary Papers
- Ryan VH, Dignon GL, Zerze GH, Chabata CV, Silva R, Conicella AE, Amaya J, Burke KA, Mittal J, Fawzi NL. Mechanistic View of hnRNPA2 Low-Complexity Domain Structure, Interactions, and Phase Separation Altered by Mutation and Arginine Methylation. Mol Cell. 2018 Feb 1;69(3):465-479.e7. Epub 2018 Jan 18 PubMed.
- Marrone L, Poser I, Casci I, Japtok J, Reinhardt P, Janosch A, Andree C, Lee HO, Moebius C, Koerner E, Reinhardt L, Cicardi ME, Hackmann K, Klink B, Poletti A, Alberti S, Bickle M, Hermann A, Pandey UB, Hyman AA, Sterneckert JL. Isogenic FUS-eGFP iPSC Reporter Lines Enable Quantification of FUS Stress Granule Pathology that Is Rescued by Drugs Inducing Autophagy. Stem Cell Reports. 2018 Feb 13;10(2):375-389. Epub 2018 Jan 18 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Modena and Reggio Emilia
The finding by Veronica H. Ryan and colleagues that wild-type and disease-linked mutated hnRNPA2 promote the phase separation of other low-complexity proteins, such as TDP-43, is important because it suggests that co-phase separation of these proteins could potentially converge, in a plethora of diseases, onto a common pathomechanism: conversion of dynamic membrane-less condensates into irreversible protein aggregates. This conversion into protein aggregates is enhanced by the defective clearance of misfolded proteins and by a general failure of the protein folding and degradation machineries, which has also been linked to aging and neurodegeneration. In agreement, Marrone and colleagues report that drugs that induce autophagy rescue FUS stress-granule pathology in iPS cell and fruit-fly models, most likely by removing misfolded proteins that would otherwise co-aggregate with stress granules. Based on these data, drugs that promote the degradation of misfolded proteins and thus prevent the conversion of stress granules into aggregates hold great promise for the development of therapeutic approaches for several types of age-related neurodegenerative diseases, with different symptoms, ranging from cognitive to motor defects.
St. Jude Children's Research Hospital
Ryan and colleagues make an important observation, and conceptually the same as we observed in our recent Neuron paper in which ALS/FTD-causing mutations were found to alter the biophysical properties of TIA-1 (Mackenzie et al., 2017). Disease-causing mutations in hnRNPA1, hnRNPA2, and TIA-1 strengthen the cohesive forces that give rise to phase transitions, and change the material properties of the liquid phase. Not addressed in the Fawzi paper is the impact on intracellular phase transitions, but the prediction is that it would have the same impact as TIA-1 mutations, which impair the dynamics of RNA granules. Indeed, in our initial paper describing disease mutations in hnRNPA1 and hnRNPA2 we reported that poorly dynamic stress granules containing these RNA-binding proteins accumulate in patient-derived cells. These same changes increase the propensity of the protein to assemble into a fibrillar “aggregate” similar to that observed in disease. It remains to be determined which is more important for driving cellular dysfunction: altered material properties (and therefore function) of the RNA granules that contain mutant hnRNP (hnRNPA1, TIA-1, hnRNPA2, TDP-43, FUS), or acquisition of a toxic property by the aggregated form of the protein. I favor the former, but time will tell. The fact that these phase transitions are regulated by posttranslational modifications is also very important, and suggests strategies for finding small molecules to restore normal RNA granule dynamics.
The Marrone et al. paper is conceptually consistent with the observation that poorly dynamic RNA granules (e.g., stress granules) that arise from mutations in hnRNPs are cleared by autophagy, otherwise known as granulophagy (Buchan et al., 2013), and underscore the potential for this process to be targeted for therapeutic benefit.
References:
Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, Matchett BJ, Mittag T, Temirov J, Hsiung GR, Krieger C, Murray ME, Kato M, Fryer JD, Petrucelli L, Zinman L, Weintraub S, Mesulam M, Keith J, Zivkovic SA, Hirsch-Reinshagen V, Roos RP, Züchner S, Graff-Radford NR, Petersen RC, Caselli RJ, Wszolek ZK, Finger E, Lippa C, Lacomis D, Stewart H, Dickson DW, Kim HJ, Rogaeva E, Bigio E, Boylan KB, Taylor JP, Rademakers R. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron. 2017 Aug 16;95(4):808-816.e9. PubMed.
Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. 2013 Jun 20;153(7):1461-74. PubMed.
Brown University
Marrone et al. describe the creation of interesting cell reporters expressing GFP-tagged forms of wild-type and disease-associated mutants of FUS. Especially interesting is the creation of closely related lines that link GFP (a large modification) with two different length linkers to the C-terminal end of FUS. Noticing that the short link alters stress granule phenotype compared to the long linker, the authors take advantage of both linker lines for designing assays optimized for sensitivity. They use these cells to show that stimulating autophagy decreases FUS accumulation in stress granules and identifies currently FDA-approved compounds for modulating.
The results will be exciting to test in recent and upcoming cell and animal models of ALS. Of further interest is the examination of a FUS variant that disturbs the canonical FUS binding site for nuclear import protein karyopherin beta2 yet does not entirely disrupt nuclear localization and redistribution after stress. These findings suggests that work on the roles of transportin/karyopherin FUS interactions and on SGs is of keen interest.
JGU Mainz
Ryan et al. used NMR spectroscopy, molecular simulations, and biochemical LLPS assays to characterize the structural details and phase-separation properties of the hnRNP-A2 low-complexity (LC) domain. A particularly interesting finding of this work is that genetic mutations in the hnRNP-A2 LC domain that are linked to neurodegenerative disease (D290V and P298L) enhance self-interaction of the LC domain and thus promote a liquid-to-solid-state transition (i.e. aggregation) of the hnRNP-A2 LC domain. Similar findings have been reported previously for disease-linked mutations in the LC domain of other RNA-binding proteins, e.g. FUS (Patel et al., 2015; Murakami et al., 2015) or hnRNP-A1 (Molliex et al., 2015), hence this seems to be a common theme in neurodegenerative diseases. The detrimental downstream changes in cells elicited by such aberrantly self-interacting and aggregating mutant RNA-binding proteins remain to be clarified. Do they promote co-phase separation and aggregation of other LC domain-containing RNA-binding proteins, such as TDP-43? Is alternative splicing of hnRNP-A2 target altered due to enhanced self-interactions of hnRNP-A2 genes (Martinez el al., 2016)? These are interesting questions for follow-up studies sparked by the present study by Fawzi and colleagues.
Another interesting finding reported by Ryan et al. is that arginine methylation on four sites reduces LLPS of the hnRNP-A2 LC domain. This highlights that posttranslational modifications in LC domains are crucial factors influencing LLPS, as previously reported for other RNA-binding proteins (Nott et al., 2015; Monahan et al., 2017), so they are promising targets for modulating LLPS/aggregation of disease-linked LC domain-containing RNA-binding proteins. Arginine methylation seems to be a particularly interesting modification, since it is often mis-regulated in disease (Thandapani et al., 2015) and arginine methylation defects have been identified in FUS-associated neurodegeneration (Dormann et al., 2012; Suárez-Calvet et al., 2016). Thus, arginine methylation could indeed have a wider role in preventing LLPS and aggregation of LC domain-containing RNA-binding proteins.
References:
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, Pozniakovski A, Poser I, Maghelli N, Royer LA, Weigert M, Myers EW, Grill S, Drechsel D, Hyman AA, Alberti S. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 2015 Aug 27;162(5):1066-77. PubMed.
Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH, Kronenberg-Versteeg D, Li Y, Yang SP, Wakutani Y, Meadows W, Ferry RR, Dong L, Tartaglia GG, Favrin G, Lin WL, Dickson DW, Zhen M, Ron D, Schmitt-Ulms G, Fraser PE, Shneider NA, Holt C, Vendruscolo M, Kaminski CF, St George-Hyslop P. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron. 2015 Nov 18;88(4):678-90. Epub 2015 Oct 29 PubMed.
Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015 Sep 24;163(1):123-33. PubMed.
Martinez FJ, Pratt GA, Van Nostrand EL, Batra R, Huelga SC, Kapeli K, Freese P, Chun SJ, Ling K, Gelboin-Burkhart C, Fijany L, Wang HC, Nussbacher JK, Broski SM, Kim HJ, Lardelli R, Sundararaman B, Donohue JP, Javaherian A, Lykke-Andersen J, Finkbeiner S, Bennett CF, Ares M Jr, Burge CB, Taylor JP, Rigo F, Yeo GW. Protein-RNA Networks Regulated by Normal and ALS-Associated Mutant HNRNPA2B1 in the Nervous System. Neuron. 2016 Nov 23;92(4):780-795. Epub 2016 Oct 20 PubMed.
Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD, Baldwin AJ. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell. 2015 Mar 5;57(5):936-47. PubMed.
Monahan Z, Ryan VH, Janke AM, Burke KA, Rhoads SN, Zerze GH, O'Meally R, Dignon GL, Conicella AE, Zheng W, Best RB, Cole RN, Mittal J, Shewmaker F, Fawzi NL. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017 Oct 16;36(20):2951-2967. Epub 2017 Aug 8 PubMed.
Thandapani P, O'Connor TR, Bailey TL, Richard S. Defining the RGG/RG motif. Mol Cell. 2013 Jun 6;50(5):613-23. PubMed.
Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C, Kremmer E, Ansorge O, Mackenzie IR, Neumann M, Haass C. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012 Sep 11; PubMed.
Suárez-Calvet M, Neumann M, Arzberger T, Abou-Ajram C, Funk E, Hartmann H, Edbauer D, Kremmer E, Göbl C, Resch M, Bourgeois B, Madl T, Reber S, Jutzi D, Ruepp MD, Mackenzie IR, Ansorge O, Dormann D, Haass C. Monomethylated and unmethylated FUS exhibit increased binding to Transportin and distinguish FTLD-FUS from ALS-FUS. Acta Neuropathol. 2016 Apr;131(4):587-604. Epub 2016 Feb 19 PubMed.
Make a Comment
To make a comment you must login or register.