Janus-Faced PLCγ2? Alzheimer’s Risk Protein Toggles TREM2 and TLR Pathways
Quick Links
Rare variants in TREM2 and PLCG2 influence a person’s odds of developing Alzheimer’s disease, but that is far from all the two genes have in common. According to a study published June 8 in Nature Neuroscience, phospholipase C γ2 acts downstream of TREM2 in a signaling pathway that supports critical microglial functions. Using human microglia derived from induced pluripotent stem cells, researchers led by Joseph Lewcock at Denali Therapeutics in South San Francisco reported that knocking out either gene product prevented the immune cells from efficiently processing lipids and neuronal debris. The researchers also found that, independently of TREM2, PLCγ2 is involved in a pro-inflammatory side hustle dictated by toll-like receptors, which, it so happens, is exacerbated by intracellular lipid build-up. Taken together, the findings strongly implicate faulty microglial lipid handling in the etiology of AD, and support therapeutic strategies that aim to rev up TREM2 signaling.
- AD risk genes TREM2 and PLCγ2 form part of same signaling pathway.
- PLCγ2 helps microglia clear engulfed debris and survive.
- Separately, PLCγ2 also acts downstream of toll-like receptors to stoke inflammation.
“Using an impressive array of experimental conditions in gene-edited iPSC-microglia, [the authors] demonstrate that PLCγ2 is a downstream effector of TREM2 and a regulator of lipid metabolism. This exciting discovery directly connects PLCγ2 to well-established AD pathways involving APOE, TREM2, and microglial activation,” commented Rik van der Kant, Vrije University, Amsterdam (full comment below). Florent Ginhoux of the Agency for Science, Technology and Research in Singapore, agreed. “The study elegantly links TREM2 and PLCγ2 signaling pathways, and offers mechanistic insight into how variants in these genes affect the pathophysiology of AD,” Ginhoux wrote (full comment below).

Double Dealing. When triggered by TREM2, PLCγ2 supports lipid metabolism and survival (left). When triggered by TLRs, PLCγ2 triggers inflammation. In TREM2 KO microglia (right), lipids accumulate and this exacerbates the pro-inflammatory, TLR-driven pathway. [Courtesy of Andreone et al., Nature Neuroscience, 2020.]
Since the discovery, in 2012, that rare variants in the coding region of TREM2 triple the risk of AD, researchers have pegged the receptor as supporting myriad microglial functions, including phagocytosis, walling off Aβ plaques, and promoting an anti-inflammatory, neuroprotective environment (May 2016 news; Apr 2017 conference news; Jul 2018 conference news).
Separately, researchers discovered a rare variant in phospholipase C γ2 (PLCG2) that protects against AD (Aug 2017 conference news on Sims et al., 2017). PLCs are a large family of intracellular enzymes that cleave the membrane phospholipid phosphatidylinositol-4,5-bisphosphate (PIP2) to diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3), a process that facilitates calcium signaling. In the brain, the γ2 isoform is predominantly expressed by microglia, and initial studies suggest that the protective variant munches phospholipids with more gusto than the common one does (Zhang et al., 2014; May 2019 news).
Might the functions of TREM2 and PLCγ2 intersect in microglia? To study this question, co-first authors Benjamin Andreone and Laralynne Przybyla derived human microglia. They wove together elements from three recently developed protocols to coax so-called induced microglia (iMGs) from induced pluripotent stem cells (Muffat et al., 2016; Pandya et al., 2017; McQuade et al., 2018). They then used CRISPR to wipe out expression of TREM2 or PLCG2 in these cell-based models.
Under normal conditions, iMGs missing either TREM2 or PLCG2 appeared healthy and viable. When the going got tough—i.e., when growth factors were depleted from the culture media—both types of knockout suffered a similar fate, dying sooner than their wild-type counterparts. The transcriptomes of each of the two iMG knockouts also differed from those of wild-type cells in similar ways. Specifically, half of the genes differentially expressed in TREM2 KO iMGs were similarly affected in PLCG KO iMGs. These common genes were part of signal transduction pathways downstream of DAP12, the adaptor protein that mediates TREM2 signaling. Using biochemical approaches, the researchers ultimately pieced together a signaling cascade by which lipids activate TREM2, leading to the phosphorylation of Syk2, which directly interacts with PLCγ2, unleashing its phospholipase activity and downstream signaling events.
Disabling the pathway, either by knocking out TREM2 or PLCγ2, had a dramatic impact on the processing of lipids, including cholesterol-laden myelin. All microglial lines in this study readily engulfed this type of fluorescently labeled debris; however, while wild-type cells had largely disposed of it after four days, TREM2 or PLCG2 knockouts were still chock-full of it by then. Tellingly, perhaps, the knockout cells failed to ramp up expression of several lipid processing genes in response to the myelin challenge.
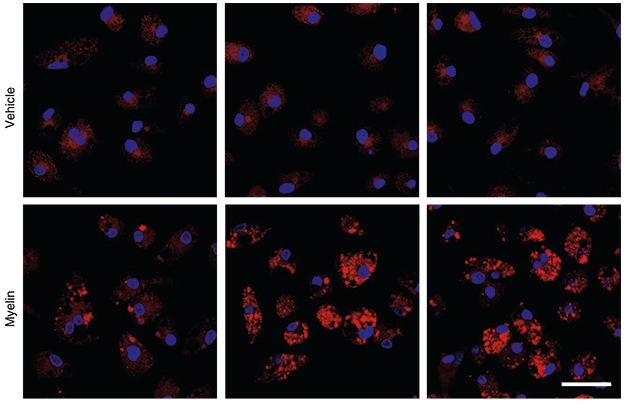
Choking on Lipids? Wild-type microglia (left) readily digested lipids after treatment with myelin, while microglia lacking PLCG2 (middle) and TREM2 (right) accumulated the lipids. [Courtesy of Andreone et al., Nature Neuroscience, 2020.]
Lipidomics experiments revealed that the knockouts became burdened with a backlog of several subtypes of unprocessed lipid, including free cholesterol, cholesteryl esters, and myelin-derived ceramides. Similarly, in co-culture experiments with iPSC-derived neurons, both types of microglial knockout were unable to properly digest detritus from injured axons.
How might AD risk variants shift these phenotypes? The researchers generated iMGs that expressed the R47H variant of TREM2, or the protective P522R variant of PLCG2. As might be expected from prior findings on these variants, the R47H-TREM2 iMGs processed lipids more sluggishly than wild-type, whereas the P522R-PLCG2 microglia more deftly disposed of them than wild-type. Together, the findings support the idea that TREM2 and PLCG2 variants influence AD risk via lipid metabolism.
Lest a reader be tempted to tie a neat little bow on this set of results, here comes the twist: PLCγ2 also takes marching orders from toll-like receptors. This was previously reported in peripheral immune cells. The Denali researchers found the same in iMGs, as PLCG2 knockouts failed to mount a pro-inflammatory response to the TLR2 ligand zymosan.
Interestingly, the same pro-inflammatory cytokines that were down in response to zymosan in PLCG2 knockout iMGs were up in TREM2 knockout iMGs. For example, compared with wild-type iMGs treated with zymosan, PLCG2 knockouts secreted 50 percent less IL-1β, while TREM2 knockouts secreted 64 percent more.
The same pattern emerged when the researchers used the TLR4 ligand LPS to trigger the microglial NLRP3 inflammasome, which itself has been tied to AD (Nov 2019 news). Loading up the microglia with myelin prior to triggering the inflammasome dramatically enhanced the inflammatory response in TREM2 KO iMGs, the scientists report. This implies that intracellular lipid accumulation may exacerbate damaging inflammatory pathways. The findings dovetail with those of a recent study that tied lipid droplet-accumulating microglia (LAM) in the aging hippocampus to neuroinflammation (Aug 2019 news).
Overall, the findings cast PLCγ2 as a two-faced player in microglia. When triggered via TREM2, this phospholipase facilitates processing of lipids and microglial survival. When tripped off by TLRs, it ramps up potentially damaging pro-inflammatory responses. And when lipids build up, as might occur in the aging brain, they exacerbate the pro-inflammatory pathway, Andreone told Alzforum. He believes the balance between these two PLCγ2 signaling pathways could dictate whether microglia help or harm.
The findings lend support to a therapeutic strategy of agonizing TREM2 signaling, Lewcock told Alzforum. That the protective PLCγ2 variant enhances lipid processing in microglia fits with the idea that even people whose TREM2 functions normally could stand to benefit from a boost in this pathway. Activating PLCγ2 is also a potential strategy, Lewcock said, although it would come with the risk of rousing its pro-inflammatory side. More work is needed to dissect how the PLCγ2 protective variant influences signaling downstream of TREM2 versus TLRs.
“This is a very important paper,” wrote Christian Haass at the German Center for Neurodegenerative Diseases in Munich. Haass noted that its findings fit with fresh data from his and other groups, but also cautioned that the molecular signature of a protective subpopulation of microglia needs to be defined in much greater detail (full comment below).
Denali is collaborating with Haass’ group to develop an activating antibody for TREM2, which will come with a blood-brain barrier transport vehicle to shuttle it into the brain (May 2019 conference news; May 2020 news). AL002, a TREM2-activating antibody developed by Alector and Abbvie, entered early clinical trials last year (see clinicaltrials.gov).—Jessica Shugart
References
News Citations
- Barrier Function: TREM2 Helps Microglia to Compact Amyloid Plaques
- New Evidence Confirms TREM2 Binds Aβ, Drives Protective Response
- TREM2: Diehard Microglial Supporter, Consequences Be DAMed
- Searching for New AD Risk Variants? Move Beyond GWAS
- The Mutation You Want: It Protects the Brain, Extends Life
- Microglia Inflammasome Stokes Tau Phosphorylation, Tangles
- Newly Identified Microglia Contain Lipid Droplets, Harm Brain
- Antibodies Against Microglial Receptors TREM2 and CD33 Head to Trials
- Molecular Transport Vehicle Shuttles Therapies into Brain
Therapeutics Citations
Paper Citations
- Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC, Martin ER, Grenier-Boley B, Heilmann-Heimbach S, Chouraki V, Kuzma AB, Sleegers K, Vronskaya M, Ruiz A, Graham RR, Olaso R, Hoffmann P, Grove ML, Vardarajan BN, Hiltunen M, Nöthen MM, White CC, Hamilton-Nelson KL, Epelbaum J, Maier W, Choi SH, Beecham GW, Dulary C, Herms S, Smith AV, Funk CC, Derbois C, Forstner AJ, Ahmad S, Li H, Bacq D, Harold D, Satizabal CL, Valladares O, Squassina A, Thomas R, Brody JA, Qu L, Sánchez-Juan P, Morgan T, Wolters FJ, Zhao Y, Garcia FS, Denning N, Fornage M, Malamon J, Naranjo MC, Majounie E, Mosley TH, Dombroski B, Wallon D, Lupton MK, Dupuis J, Whitehead P, Fratiglioni L, Medway C, Jian X, Mukherjee S, Keller L, Brown K, Lin H, Cantwell LB, Panza F, McGuinness B, Moreno-Grau S, Burgess JD, Solfrizzi V, Proitsi P, Adams HH, Allen M, Seripa D, Pastor P, Cupples LA, Price ND, Hannequin D, Frank-García A, Levy D, Chakrabarty P, Caffarra P, Giegling I, Beiser AS, Giedraitis V, Hampel H, Garcia ME, Wang X, Lannfelt L, Mecocci P, Eiriksdottir G, Crane PK, Pasquier F, Boccardi V, Henández I, Barber RC, Scherer M, Tarraga L, Adams PM, Leber M, Chen Y, Albert MS, Riedel-Heller S, Emilsson V, Beekly D, Braae A, Schmidt R, Blacker D, Masullo C, Schmidt H, Doody RS, Spalletta G, Jr WT, Fairchild TJ, Bossù P, Lopez OL, Frosch MP, Sacchinelli E, Ghetti B, Yang Q, Huebinger RM, Jessen F, Li S, Kamboh MI, Morris J, Sotolongo-Grau O, Katz MJ, Corcoran C, Dunstan M, Braddel A, Thomas C, Meggy A, Marshall R, Gerrish A, Chapman J, Aguilar M, Taylor S, Hill M, Fairén MD, Hodges A, Vellas B, Soininen H, Kloszewska I, Daniilidou M, Uphill J, Patel Y, Hughes JT, Lord J, Turton J, Hartmann AM, Cecchetti R, Fenoglio C, Serpente M, Arcaro M, Caltagirone C, Orfei MD, Ciaramella A, Pichler S, Mayhaus M, Gu W, Lleó A, Fortea J, Blesa R, Barber IS, Brookes K, Cupidi C, Maletta RG, Carrell D, Sorbi S, Moebus S, Urbano M, Pilotto A, Kornhuber J, Bosco P, Todd S, Craig D, Johnston J, Gill M, Lawlor B, Lynch A, Fox NC, Hardy J, ARUK Consortium, Albin RL, Apostolova LG, Arnold SE, Asthana S, Atwood CS, Baldwin CT, Barnes LL, Barral S, Beach TG, Becker JT, Bigio EH, Bird TD, Boeve BF, Bowen JD, Boxer A, Burke JR, Burns JM, Buxbaum JD, Cairns NJ, Cao C, Carlson CS, Carlsson CM, Carney RM, Carrasquillo MM, Carroll SL, Diaz CC, Chui HC, Clark DG, Cribbs DH, Crocco EA, DeCarli C, Dick M, Duara R, Evans DA, Faber KM, Fallon KB, Fardo DW, Farlow MR, Ferris S, Foroud TM, Galasko DR, Gearing M, Geschwind DH, Gilbert JR, Graff-Radford NR, Green RC, Growdon JH, Hamilton RL, Harrell LE, Honig LS, Huentelman MJ, Hulette CM, Hyman BT, Jarvik GP, Abner E, Jin LW, Jun G, Karydas A, Kaye JA, Kim R, Kowall NW, Kramer JH, LaFerla FM, Lah JJ, Leverenz JB, Levey AI, Li G, Lieberman AP, Lunetta KL, Lyketsos CG, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Morris JC, Murrell JR, Myers AJ, O'Bryant S, Olichney JM, Pankratz VS, Parisi JE, Paulson HL, Perry W, Peskind E, Pierce A, Poon WW, Potter H, Quinn JF, Raj A, Raskind M, Reisberg B, Reitz C, Ringman JM, Roberson ED, Rogaeva E, Rosen HJ, Rosenberg RN, Sager MA, Saykin AJ, Schneider JA, Schneider LS, Seeley WW, Smith AG, Sonnen JA, Spina S, Stern RA, Swerdlow RH, Tanzi RE, Thornton-Wells TA, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Van Eldik LJ, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Wilhelmsen KC, Williamson J, Wingo TS, Woltjer RL, Wright CB, Yu CE, Yu L, Garzia F, Golamaully F, Septier G, Engelborghs S, Vandenberghe R, De Deyn PP, Fernadez CM, Benito YA, Thonberg H, Forsell C, Lilius L, Kinhult-Stählbom A, Kilander L, Brundin R, Concari L, Helisalmi S, Koivisto AM, Haapasalo A, Dermecourt V, Fievet N, Hanon O, Dufouil C, Brice A, Ritchie K, Dubois B, Himali JJ, Keene CD, Tschanz J, Fitzpatrick AL, Kukull WA, Norton M, Aspelund T, Larson EB, Munger R, Rotter JI, Lipton RB, Bullido MJ, Hofman A, Montine TJ, Coto E, Boerwinkle E, Petersen RC, Alvarez V, Rivadeneira F, Reiman EM, Gallo M, O'Donnell CJ, Reisch JS, Bruni AC, Royall DR, Dichgans M, Sano M, Galimberti D, St George-Hyslop P, Scarpini E, Tsuang DW, Mancuso M, Bonuccelli U, Winslow AR, Daniele A, Wu CK, GERAD/PERADES, CHARGE, ADGC, EADI, Peters O, Nacmias B, Riemenschneider M, Heun R, Brayne C, Rubinsztein DC, Bras J, Guerreiro R, Al-Chalabi A, Shaw CE, Collinge J, Mann D, Tsolaki M, Clarimón J, Sussams R, Lovestone S, O'Donovan MC, Owen MJ, Behrens TW, Mead S, Goate AM, Uitterlinden AG, Holmes C, Cruchaga C, Ingelsson M, Bennett DA, Powell J, Golde TE, Graff C, De Jager PL, Morgan K, Ertekin-Taner N, Combarros O, Psaty BM, Passmore P, Younkin SG, Berr C, Gudnason V, Rujescu D, Dickson DW, Dartigues JF, DeStefano AL, Ortega-Cubero S, Hakonarson H, Campion D, Boada M, Kauwe JK, Farrer LA, Van Broeckhoven C, Ikram MA, Jones L, Haines JL, Tzourio C, Launer LJ, Escott-Price V, Mayeux R, Deleuze JF, Amin N, Holmans PA, Pericak-Vance MA, Amouyel P, van Duijn CM, Ramirez A, Wang LS, Lambert JC, Seshadri S, Williams J, Schellenberg GD. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer's disease. Nat Genet. 2017 Sep;49(9):1373-1384. Epub 2017 Jul 17 PubMed.
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014 Sep 3;34(36):11929-47. PubMed.
- Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, Bakiasi G, Tsai LH, Aubourg P, Ransohoff RM, Jaenisch R. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med. 2016 Nov;22(11):1358-1367. Epub 2016 Sep 26 PubMed.
- Pandya H, Shen MJ, Ichikawa DM, Sedlock AB, Choi Y, Johnson KR, Kim G, Brown MA, Elkahloun AG, Maric D, Sweeney CL, Gossa S, Malech HL, McGavern DB, Park JK. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat Neurosci. 2017 May;20(5):753-759. Epub 2017 Mar 2 PubMed.
- McQuade A, Coburn M, Tu CH, Hasselmann J, Davtyan H, Blurton-Jones M. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol Neurodegener. 2018 Dec 22;13(1):67. PubMed.
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Andreone BJ, Przybyla L, Llapashtica C, Rana A, Davis SS, van Lengerich B, Lin K, Shi J, Mei Y, Astarita G, Di Paolo G, Sandmann T, Monroe KM, Lewcock JW. Alzheimer's-associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat Neurosci. 2020 Aug;23(8):927-938. Epub 2020 Jun 8 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
cclaes@uci.edu
University of California, Irvine
This exciting new study by Andreone et al. examines the importance of PLCy2 activity downstream of TREM2 signaling for survival, myelin phagocytosis, lipid metabolism, and processing of neuronal debris by human microglia. This work also provides an elegant example of how iPSC-derived microglia can be used to examine the biology and interactions between AD risk genes. By combining CRISPR gene editing with iPS-microglia (iMG), Andreone et al. examine the impact of TREM2 or PLCG2 deletion on microglial gene expression, function, and lipid metabolism, providing important new insight into the role of and interactions between these two AD risk genes.
As TREM2 signaling is known to be mediated at least in part by PLCy2 activity, the authors began by comparing the transcriptional changes that occur between TREM2 and PLCy2 isogenic knockout lines. As expected, many changes are shared between these two lines, further demonstrating the importance of PLCy2 in TREM2 signaling. However, the authors also uncover many additional genes that are downregulated only in PLCy2 KO microglia. Next, to determine whether TREM2 and PLCy2 deletion confers similar functional effects, the authors examine the microglial response to myelin, demonstrating that deletion in either gene leads to the accumulation of myelin and a failure to induce key lipid metabolism genes. By performing elegant lipidomic analysis, Andreone and colleagues further show that these impaired responses also lead to a highly similar accumulation of sterol species within TREM2 and PLCy2 knockout microglia. Importantly, this failed myelin response is paralleled by a similar failure in the clearance of neuronal debris in a co-culture axotomy model. But what about the AD-associated R47H TREM2 mutation? To determine whether this mutation confers similar effects to deletion, the authors performed rescue experiments to induce TREM2 R47H expression within TREM2 knockout microglia and reveal a similar accumulation of myelin and cholesterol esters as observed in TREM2 heterozygous knockout cells, suggesting that the R47H mutation acts as a hypomorph. Using an equivalent approach to examine the PLCy2 P522R protective AD variant, the authors demonstrate that P522R further reduces microglial lipid accumulation compared with wild-type PLCy2.
So, do TREM2 and PLCy2 deletions simply phenocopy each other? As first alluded to in their initial RNA-sequencing experiment, PLCy2 deletion causes a downregulation of many additional genes that are not altered in TREM2 KO microglia. Indeed, in the periphery PLCy2 has been reported to signal downstream of multiple pro-inflammatory TLRs, thus the authors treated their iMGs with zymosan, a TLR ligand. This additional experiment showed that most zymosan-induced genes were shared between wild-type and TREM2 KO microglia, suggesting that TREM2 is not directly involved in TLR signaling. In contrast, ~50 percent fewer genes were upregulated in PLCy2 KOs treated with zymosan, supporting the notion that as in the periphery, microglial PLCy2 activity is also critical for TLR signaling. Notably, levels of IL1β, a proinflammatory cytokine that is strongly implicated in AD, were reduced by 50 percent in zymosan-treated PLCy2 KO cells, but conversely increased by 64 percent in zymosan-treated TREM2 KOs. Thus, PLCy2 signaling is required for TLR-mediated proinflammatory signaling, which under normal circumstances appears to be dampened by TREM2 activity. Additionally, NLRP3 inflammasome activation (which induces IL1β maturation) was attenuated upon PLCy2 loss, while TREM2 KOs secreted significantly more IL1β than wild-type cells. In this respect, the negative impact of microglial NLRP3 on both amyloid and tau pathology has been clearly shown (Venegas et al., 2017; Heneka et al., 2018; Ising et al., 2019). Thus, one must be cautious to explore therapies that simply increase PLCy2 signaling, as proper TREM2 function is likely also crucial to prevent an overactive hyperinflammatory state.
Overall, this is an insightful and compelling study that greatly advances our understanding of TREM2 and PLCy2 signaling in human microglia. However, there are a couple of questions and caveats that future studies will hopefully address. The authors mention that loss of either TREM2 or PLCy2 reduces phagocytosis of both myelin and neurons and induced lipid accumulation. As TREM2 is not the only receptor responsible for the uptake of these ligands, one might hypothesize that the impaired phagocytosis of myelin or neuronal debris might also be exacerbated by defective degradation of these lipid-laden substrates which could in turn feed back to alter the expression of other relevant receptors. One technical caveat to also consider is that the rescue experiments were achieved through lentiviral transduction. Our lab has found in preliminary studies that lentiviral expression of GFP alone in iMGs changes thousands of genes. Thus, we need to be cautious with the interpretation of viral manipulations in a cell type that plays a role in viral defense. Another curious finding was the detection of reduced TREM2 mRNA expression in the R47H rescue experiment. This observation contrasts with the finding that TREM2 mRNA expression and splicing is unchanged in human R47H iMGs and patient brains (Xiang et al., 2018). However, the authors confirmed similar membrane localization of wild-type and R47H TREM2 protein and thus the observed effects of the R47H mutation are likely initially induced by altered ligand binding and reduced downstream signaling.
One last caveat to consider is that in vitro iPS-microglia models most closely resemble cultured human microglia (Gosselin et al., 2017; Hasselmann et al., 2019). As significant differences exist between in vitro and in vivo microglial gene expression and perhaps then also microglial function, it will critical to follow up on this elegant work by examining similar questions in chimeric model systems (Hasselmann et al., 2019; Mancuso et. al., 2019). In this respect, ongoing studies in our lab are examining both P522R PLCy2 and TREM2 R47H mutant human microglia in chimeric AD mice. Thus far, our data indicates lipid accumulation and enrichment of many lipid-associated genes holds true in human DAMs in AD mice. Our ongoing lipidomic studies will hopefully further determine the extent to which these exciting findings hold true in AD chimeric mice in vivo.
Ongoing analysis of the impact of these mutations on microglia function, lipidomics, and AD pathology will hopefully reveal whether these risk genes influence microglia function in vivo as elegantly predicted by this important study.
References:
Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, Griep A, Gelpi E, Beilharz M, Riedel D, Golenbock DT, Geyer M, Walter J, Latz E, Heneka MT. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer's disease. Nature. 2017 Dec 20;552(7685):355-361. PubMed.
Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci. 2018 Oct;19(10):610-621. PubMed.
Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, Griep A, Santarelli F, Brosseron F, Opitz S, Stunden J, Merten M, Kayed R, Golenbock DT, Blum D, Latz E, Buée L, Heneka MT. NLRP3 inflammasome activation drives tau pathology. Nature. 2019 Nov;575(7784):669-673. Epub 2019 Nov 20 PubMed.
Xiang X, Piers TM, Wefers B, Zhu K, Mallach A, Brunner B, Kleinberger G, Song W, Colonna M, Herms J, Wurst W, Pocock JM, Haass C. The Trem2 R47H Alzheimer's risk variant impairs splicing and reduces Trem2 mRNA and protein in mice but not in humans. Mol Neurodegener. 2018 Sep 6;13(1):49. PubMed.
Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JC, Sajti E, Jaeger BN, O'Connor C, Fitzpatrick C, Pasillas MP, Pena M, Adair A, Gonda DD, Levy ML, Ransohoff RM, Gage FH, Glass CK. An environment-dependent transcriptional network specifies human microglia identity. Science. 2017 Jun 23;356(6344) Epub 2017 May 25 PubMed.
Hasselmann J, Coburn MA, England W, Figueroa Velez DX, Kiani Shabestari S, Tu CH, McQuade A, Kolahdouzan M, Echeverria K, Claes C, Nakayama T, Azevedo R, Coufal NG, Han CZ, Cummings BJ, Davtyan H, Glass CK, Healy LM, Gandhi SP, Spitale RC, Blurton-Jones M. Development of a Chimeric Model to Study and Manipulate Human Microglia In Vivo. Neuron. 2019 Sep 25;103(6):1016-1033.e10. Epub 2019 Jul 30 PubMed.
Mancuso R, Van Den Daele J, Fattorelli N, Wolfs L, Balusu S, Burton O, Liston A, Sierksma A, Fourne Y, Poovathingal S, Arranz-Mendiguren A, Sala Frigerio C, Claes C, Serneels L, Theys T, Perry VH, Verfaillie C, Fiers M, De Strooper B. Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat Neurosci. 2019 Dec;22(12):2111-2116. Epub 2019 Oct 28 PubMed.
Biomedizinisches Centrum (BMC), Biochemie & Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE)
This is a very important paper, because it supports the idea to therapeutically modulate TREM2 functions in microglia. Andreone and colleagues independently support this approach by demonstrating shared LOF phenotypes upon TREM2 or PLCγ2 knockout. This suggests that promoting TREM2/PLCγ2-dependent lipid metabolism may be protective in AD.
This also nicely fits with our recent findings, together with Mikko Hiltunen, showing that the protective mutation stimulates exactly those functions, which are reduced by the TREM2 LOF.
However, as is always the case with microglia, any beneficial change may be associated with detrimental events. In this case, this is a TREM2-independent function downstream of toll-like receptors, which mediates an inflammatory response. Therefore, we need to understand in much greater detail the molecular signature of a protective microglia sub-population, and specifically how this population can be modulated/stimulated by therapeutic anti-TREM2 antibodies.
This paper also demonstrates very nicely the fantastic potential of iMG as models for functional analyses in a human background.
Vrije Universiteit Amsterdam
This study provides important new insights into the cell biological functions of PLCγ2 in (human) microglia. It has been known for some years that a coding variant in PLCγ2 (P522R) protects from Alzheimer’s disease and other dementias, but how PLCγ2 (a phospholipase C enriched in myeloid cells) connects to established AD pathogenic pathways remained unknown and difficult to foretell.
Using an impressive array of experimental conditions in gene-edited iPSC-microglia, Andreone and Przybyla et al. demonstrate that PLCγ2 is a downstream effector of TREM2 and a regulator of lipid metabolism. This exciting discovery directly connects PLCγ2 to well-established AD pathways involving APOE, TREM2, and microglial activation. The authors show that PLCγ2 regulates microglial lipid homeostasis, lipid clearance, and is involved in lipid-dependent inflammatory signaling in human microglia, similar to what has been previously reported for TREM2 and APOE (Nugent et al., 2020).
Furthermore, the study confirms previous reports (Magno et al., 2019; Maguire et al., 2020) by showing that PLCγ2 P522R is a functional hypermorph. The authors extend these findings by showing that PLCγ2 P522R more potently reduces lipid accumulation in lipid-challenged microglia than wild-type PLCγ2. The overall findings of this paper fit with a broader body of recent literature reporting on altered cholesterol metabolism downstream of SAD-risk genes in astrocytes, microglia, and neurons. The current PLCγ2 finding is particularly exciting, as it is the first to demonstrate that AD protective factors may also play into this mechanism.
Importantly, the authors also note that PLCγ2 not only functions downstream of TREM2, but is also activated by other microglial receptors such as TLRs. To what extent protection by PLCγ2 P522R is conferred through its effects on lipid metabolism versus regulation of other immune pathways will be an interesting puzzle for coming studies. The strong overlap between lipid metabolic changes in PLCγ2 and TREM2 knockout microglia does, however, suggest that cholesterol metabolism is a common converging pathway downstream of these genes, as well as downstream of APOE risk variants.
Overall, this paper takes a great stride forward toward understanding the function of PLCγ2 in human microglia and AD, and is of great value to the research community.
References:
Magno L, Lessard CB, Martins M, Lang V, Cruz P, Asi Y, Katan M, Bilsland J, Lashley T, Chakrabarty P, Golde TE, Whiting PJ. Alzheimer's disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res Ther. 2019 Feb 2;11(1):16. PubMed.
Maguire E, Menzies GE, Phillips T, Sasner M, Williams HM, Czubala MA, Evans N, Cope EL, Sims R, Howell GR, Lloyd-Evans E, Williams J, Allen ND, Taylor PR. The Alzheimer’s disease protective P522R variant of PLCG2, consistently enhances stimulus-dependent PLCγ2 activation, depleting substrate and altering cell function. bioRxiv, April 28, 2020
Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, Lucas A, Baskaran S, Haddick PC, Lenser M, Earr TK, Shi J, Dugas JC, Andreone BJ, Logan T, Solanoy HO, Chen H, Srivastava A, Poda SB, Sanchez PE, Watts RJ, Sandmann T, Astarita G, Lewcock JW, Monroe KM, Di Paolo G. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron. 2020 Mar 4;105(5):837-854.e9. Epub 2020 Jan 2 PubMed.
Indiana University School of Medicine
Stanford University
Andreone and colleagues have provided an extensive and compelling study on the roles of the PLCγ2 in human iPSC-derived microglia-like cells. A significant outcome of this study is the validation that the findings in the human iMG cells largely recapitulate those of murine microglia.
The roles of TREM2 and PLCγ2 in microglia in AD pathogenesis have commanded substantial attention by the identification that coding variants in TREM2 and PLCγ2 are associated with AD risk. The authors report that TREM2 signals through PLCγ2 to mediate cell survival, phagocytosis, processing of neuronal debris, and lipid metabolism in microglia. They demonstrate that TREM2 and PLCγ2 share common signaling pathways and that PLCγ2 act as a major regulator of microglial functional in AD. In this regard, the experimental outcomes serve largely to reinforce the current understanding of how PLCγ2 interacts with upstream and downstream signaling elements to elicit TREM2 and TLR-driven changes in microglial phenotypes.
There are a few surprises, however. For example, TREM2 was shown to suppress proinflammatory responses in microglia in the context of TLR stimulation. These data also suggest that PLCγ2 represents the point of significant intersection of TLR and TREM2 signaling in AD. This would be consistent with an older literature on the involvement of microglial TLRs in AD pathogenesis.
Recent studies provided data arguing that the protective P522R mutation in PLCγ2 promotes beneficial function in microglia through increased PLCγ2 activity, microglial activation, and enhanced endocytic clearance (Magno et al., 2019; Maguire et al., 2020; Takalo et al., 2020). The current report is consistent with these observations and broadens our understanding of the role of PLCγ2 in lipid metabolism.
Moreover, a recent study reported a novel missense variant (M28L) in PLCγ2 confers increased risk for LOAD. PLCγ2 expression was shown to be increased in several brain regions of LOAD patients and strongly correlated with brain amyloid burden in LOAD patients and AD animal models (Tsai et al., 2020). In AD, it is unclear whether the effects of genetic variants are related to alteration in enzymatic activity, the capacity of PLCγ2 to interact with other signaling elements, or topology. These findings highlight the importance of PLCγ2 biology in the brain, but also raise the question more broadly of whether increased PLCγ2 activity favors disease exacerbation or attenuation.
References:
Magno L, Lessard CB, Martins M, Lang V, Cruz P, Asi Y, Katan M, Bilsland J, Lashley T, Chakrabarty P, Golde TE, Whiting PJ. Alzheimer's disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res Ther. 2019 Feb 2;11(1):16. PubMed.
Maguire E, Menzies GE, Phillips T, Sasner M, Williams HM, Czubala MA, Evans N, Cope EL, Sims R, Howell GR, Lloyd-Evans E, Williams J, Allen ND, Taylor PR. The Alzheimer’s disease protective P522R variant of PLCG2, consistently enhances stimulus-dependent PLCγ2 activation, depleting substrate and altering cell function. bioRxiv, April 28, 2020
Takalo M, Wittrahm R, Wefers B, Parhizkar S, Jokivarsi K, Kuulasmaa T, Mäkinen P, Martiskainen H, Wurst W, Xiang X, Marttinen M, Poutiainen P, Haapasalo A, Hiltunen M, Haass C. The Alzheimer's disease-associated protective Plcγ2-P522R variant promotes immune functions. Mol Neurodegener. 2020 Sep 11;15(1):52. PubMed.
Tsai AP, Dong C, Preuss C, Moutinho M, Lin PB, Hajicek N. PLCG2 as a Risk Factor for Alzheimer’s Disease. bioRxiv: 2020.2005.2019.104216. BioRxiv.
Gustave Roussy Institute
This is a very interesting study. It linked TREM2 and PLCG2 signaling pathways in IPSC-derived macrophages/microglial cells to get insights into the mechanisms underlying the deleterious effect of TREM mutations and the protective effect of the PLCG2 P522R variant in the pathophysiology of AD.
The authors show very elegantly (using corresponding KO IPSC macrophages and rescue experiments) that disruption of TREM2 and PLCG2, which is downstream of TREM2/DAP12 signaling pathway, lead to lipid processing dysregulation and lipid accumulation further validated by lipidomic profiling. This study also further highlights the power of using iPSC-derived macrophages/microglia as tools to better understand the biology of human macrophages and their contribution to disease.
Cardiff University
This study eloquently consolidates Magno et al., 2019, and Maguire et al., 2020, and it adds to our understanding of the interaction of TREM2 and PLCγ2 as well as the independent role of PLCγ2 in iMG. The authors present a clear hypothesis as to how these AD-associated loci may act in AD etiology and highlight the complex interplay of AD risk loci in disease.
The authors use CRISPR to knock out rather than manipulate disease-specific variation. The findings correlate with the suggested loss of function of TREM2 and the gain of function of PLCγ2, as conferred by R47H and R62H in TREM2 and the protective P522R mutation in PLCG2. However, the recent identification of the M28L risk variant in PLCG2 (Tsai et al. 2020), and the observation that PLCG2 expression levels are increased in several brain regions of individuals living with AD, suggest that elevation of PLCγ2 activity could have both beneficial and detrimental effects.
Of great interest is the articulately presented utility of an iMG model to recapitulate human disease in vitro. CRISPR targeting of specific risk variants in disease-relevant models such as iMG will hopefully further unpack the complex interplay of these and other multifaceted disease loci.
This valuable research further highlights microglia and cholesterol metabolism in AD, validating the extensive genetic and epidemiological evidence. It is exciting to observe the convergence of evidence across numerous scientific fields identifying valid therapeutic targets for AD but also emphasizing the complexity of targeting multifunctional proteins.
References:
Magno L, Lessard CB, Martins M, Lang V, Cruz P, Asi Y, Katan M, Bilsland J, Lashley T, Chakrabarty P, Golde TE, Whiting PJ. Alzheimer's disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res Ther. 2019 Feb 2;11(1):16. PubMed.
Maguire E, Menzies GE, Phillips T, Sasner M, Williams HM, Czubala MA, Evans N, Cope EL, Sims R, Howell GR, Lloyd-Evans E, Williams J, Allen ND, Taylor PR. The Alzheimer’s disease protective P522R variant of PLCG2, consistently enhances stimulus-dependent PLCγ2 activation, depleting substrate and altering cell function. bioRxiv, April 28, 2020
Tsai AP, Dong C, Preuss C, Moutinho M, Lin PB, Hajicek N. PLCG2 as a Risk Factor for Alzheimer’s Disease. bioRxiv: 2020.2005.2019.104216. BioRxiv.
ARUK UCL Drug Discovery Institute
This paper provides an exciting and important step forward in the understanding of the multiple functions of the PLCγ2 enzyme in microglia.
Although previous studies have described a key role for this PLC isoform in the regulation of peripheral immune system responses, ideas on its contribution to the brain’s innate immune system have so far remained speculative. This is partly due to PLCγ2 being only recently recognized as playing a role in central neurodegenerative conditions, and to this enzyme’s involvement in complex and not fully characterized microglia signaling pathways, many of which display crosstalk with each other. Thus, it is conceivable that activation of PLCγ2 via different membrane receptors, but also via intracellular enzymes such as Rac, might contribute to several and diverse microglia functions.
Interestingly, the authors show that PLCγ2 signals not only downstream of Trem2, to support cholesterol metabolism, but also—and independently—downstream of TLR, to elicit inflammatory responses. Therefore, as discussed by the authors, activation of PLCγ2 could have opposing consequences in terms of beneficial and detrimental effects in neurodegenerative disorders, such as late-onset Alzheimer’s disease.
The DAM phenotype, which depends on the ApoE-Trem2 pathway and shows a transcriptomic profile associated with increased expression of inflammatory factors, has been associated with both beneficial and detrimental effects (Keren-Shaul et al., 2017; Krasemann et al., 2017). This might be both cell-status-dependent and dosage-dependent, as exemplified by the effect of the P522R PLCγ2 variant (Magno et al., 2019; Maguire et al., 2020; Takalo et al., 2020; this paper). Therefore, a potentially more nuanced and less dualistic interpretation of these mechanisms in AD would be needed.
Also, further investigations would be required to define the interplay between lipid metabolism and neuroinflammation, and how the key enzyme PLCγ2 is wired in the signalling networks supporting those functions.
Furthermore, it would be interesting to understand:
A selective pharmacological tool for PLCγ2 would help address some of these questions, and help define further roles of this enzyme in innate immune cell signalling pathways.
Dysregulation of lipid metabolism in the brain has been linked to Alzheimer’s disease. Andreone and colleagues neatly show that potentiation of the Trem2-PLCγ2 pathway should be further explored as a potential avenue for the development of disease-modifying therapies.
References:
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I. A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease. Cell. 2017 Jun 15;169(7):1276-1290.e17. Epub 2017 Jun 8 PubMed.
Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O'Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, Budnik B, Lemere C, Ikezu T, Heppner FL, Litvak V, Holtzman DM, Lassmann H, Weiner HL, Ochando J, Haass C, Butovsky O. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017 Sep 19;47(3):566-581.e9. PubMed.
Maguire E, Menzies GE, Phillips T, Sasner M, Williams HM, Czubala MA, Evans N, Cope EL, Sims R, Howell GR, Lloyd-Evans E, Williams J, Allen ND, Taylor PR. The Alzheimer’s disease protective P522R variant of PLCG2, consistently enhances stimulus-dependent PLCγ2 activation, depleting substrate and altering cell function. bioRxiv, April 28, 2020
Magno L, Lessard CB, Martins M, Lang V, Cruz P, Asi Y, Katan M, Bilsland J, Lashley T, Chakrabarty P, Golde TE, Whiting PJ. Alzheimer's disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res Ther. 2019 Feb 2;11(1):16. PubMed.
Takalo M, Wittrahm R, Wefers B, Parhizkar S, Jokivarsi K, Kuulasmaa T, Mäkinen P, Martiskainen H, Wurst W, Xiang X, Marttinen M, Poutiainen P, Haapasalo A, Hiltunen M, Haass C. The Alzheimer's disease-associated protective Plcγ2-P522R variant promotes immune functions. Mol Neurodegener. 2020 Sep 11;15(1):52. PubMed.
Tsai AP, Dong C, Preuss C, Moutinho M, Lin PB, Hajicek N. PLCG2 as a Risk Factor for Alzheimer’s Disease. bioRxiv: 2020.2005.2019.104216. BioRxiv.
Make a Comment
To make a comment you must login or register.