New Role for α-Synuclein: Stabilizing mRNA in P-Bodies
Quick Links
Much of the research into α-synuclein, the major component of Lewy bodies in Parkinson’s and other neurodegenerative diseases, focuses on its role in synaptic vesicles and membranes. But could other functions be relevant to disease? In the June 9 Cell, researchers led by Vikram Khurana at Brigham and Women’s Hospital, Boston, suggest as much, describing an unexpected role for α-synuclein in cytosolic mRNA processing bodies, a.k.a. P-bodies.
- α-Synuclein binds P-body proteins, disrupting these mRNA-processing organelles.
- Synuclein stabilizes transcripts, slowing their degradation.
- Genetic variants that impair P-body proteins associate with Parkinson’s disease.
In cultured human neurons, α-synuclein slowed mRNA degradation by physically interacting with key P-body proteins tasked with dismantling transcripts. In postmortem tissue from human frontal cortex, aggregates of α-synuclein correlated with mRNA accumulation. Moreover, genetic analysis linked variants in P-body genes to PD risk. Khurana believes the data could shed new light on how the disease starts in some patients, which could, in turn, guide the development of therapeutics.
Others praised the work. “It is a tour de force that harnesses the power of model systems to pinpoint an unanticipated connection between α-synuclein and P-bodies, which may have implications for PD,” James Shorter at the University of Pennsylvania, Philadelphia, wrote to Alzforum. Tim Bartels at University College London called the findings convincing. “I think the paper will be a seminal publication in the field,” Bartels wrote (full comment below).
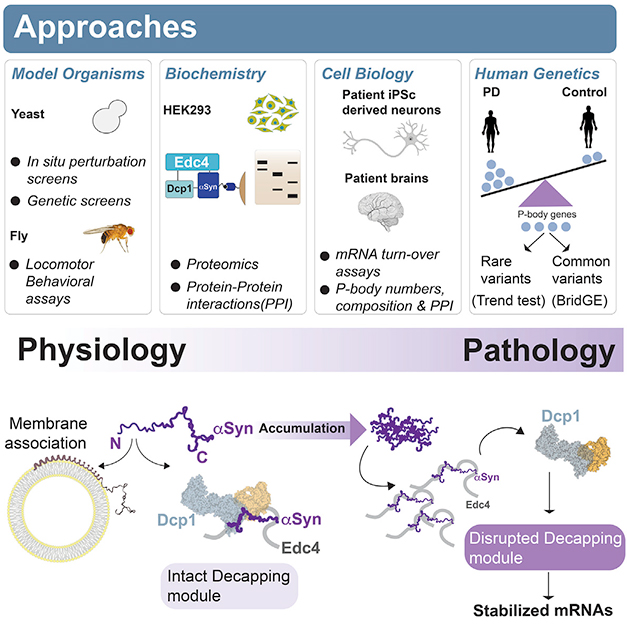
New Path to Toxicity. Normal α-synuclein (purple) can associate via its N-terminus with either membranes or with mRNA degradation proteins such as Edc4 and Dcp1. Too much α-synuclein disrupts de-capping of mRNA, leading to its buildup. [Courtesy of Hallacli et al., Cell.]
Previous work on α-synuclein has tied the protein to vesicle trafficking and fusion, especially at synapses (Jun 2010 news; Oct 2014 news; Oct 2016 news). However, while working in the late Susan Lindquist’s lab at the Whitehead Institute for Biomedical Research in Cambridge, Massachusetts, Khurana turned up connections to mRNA translation and processing, as well (Feb 2017 news). How α-synuclein affected these processes was unknown.
To follow up, first author Erinc Hallacli expressed α-synuclein in a yeast model system containing tagged RNA-binding proteins that acted as reporters. α-Synuclein affected the function of several RBPs, particularly those that regulated mRNA degradation. To pin down how α-synuclein did this, the scientists turned to a fly model of synucleinopathy. A genetic screen found that knockdown of nine different P-body genes worsened α-synuclein toxicity and impaired the flies’ ability to move. A particularly strong effect came from knockdown of the ribonuclease Xrn1, which chops up mRNA. Again, this suggested an interaction between α-synuclein and mRNA degradation genes.
Was this interaction direct? To investigate, the authors immunoprecipitated α-synuclein from HEK293 cells and identified 29 bound proteins by mass spectrometry. Four of them were P-body proteins, with the strongest interactor being Edc4. This scaffold protein helps assemble the complex responsible for snipping off the 5' cap on mRNA, allowing Xrn1 to chew up the remainder. The other three—Edc3, Dcp1, and Dcp2—are members of this de-capping complex as well.
Digging deeper, the authors found that α-synuclein associated only with small soluble P-bodies, not with the larger macro P-bodies that scientists typically study. Intriguingly, some evidence suggests that larger P-bodies are specialized to store mRNA, while the smaller bodies degrade it (Corbet and Parker, 2019). The findings help explain how a-synuclein’s association with these structures has gone undetected.
Next, the authors used human neurons generated from iPSCs to investigate α-synuclein’s function in P-bodies. The iPSCs were made from familial PD patients carrying four copies of the α-synuclein gene. In these neurons, which expressed twice the normal amount of α-synuclein, more of the protein bound to Edc4. The more α-synuclein there was, the less Edc4 interacted with Dcp1, suggesting interference with normal P-body function. Supporting this, neurons with twice the normal amount of α-synuclein had only half as many macro P-bodies as did control neurons, and they degraded mRNA more slowly. The authors noted that larger P-bodies may be in equilibrium with smaller ones. Several of the most-affected transcripts, such as SCARB2, VPS13A, and PIKFYVE, have been linked previously to PD and other neurodegenerative diseases (Jul 2011 news; Feb 2018 news; Apr 2021 news).
Likewise, in postmortem dorsolateral prefrontal cortex samples from the ROSMAP observational cohort, the more Lewy bodies there were, the more these same P-body transcripts had accumulated. In brain tissue from people who carried an α-synuclein A53T variant, or a gene duplication, the more α-synuclein-Edc4 interactions there were, the fewer Edc4-Dcp1 interactions.
Genetic analyses strengthened the case that these changes in P-bodies are relevant to disease. Parkinson’s GWAS had previously implicated one P-body gene, LSM7 (Chang et al., 2017). The authors fished for others by examining the cumulative effect of mutations in P-body genes. They found a link between these mutations and PD in seven different case-control cohorts. In a separate analysis, people with PD were likelier to carry rare variants that were predicted to impair the function of P-body genes than were healthy controls, but had the same number of silent variants in these genes as did controls. Khurana noted the finding should be corroborated in larger datasets.
How do these findings fit with α-synuclein function in synaptic vesicles? α-Synuclein binds vesicular membranes via its N-terminus, which assumes a corkscrew shape in the hydrophobic environment of the lipid bilayer. Curiously, the N-terminus is also responsible for binding de-capping proteins in the P-body. When the authors mutated α-synuclein in cultured cells to make the protein more hydrophobic and promote membrane binding, it disappeared from P-bodies. The data hint that membranes and P-bodies may compete to bind α-synuclein, creating a kind of intracellular crosstalk between these compartments.
Bartels found this illuminating. His lab and others have linked α-synuclein’s propensity to bind membranes to its toxicity, but the mechanism was unclear. “This study would imply that aberrant lipid binding does not actually [cause toxicity] via the membrane, but by preventing proper α-synuclein-P-body interaction,” Bartels noted. “If true, there will be a plethora of new exploratory studies … potentially starting a completely new field of α-synuclein biology.”
Hallacli noted that this previously unrecognized function of α-synuclein in P-bodies could complicate therapeutic strategies under consideration that would prevent the protein from clustering on membranes and impairing vesicle transport. If too much α-synuclein ends up in cytosol instead, it could impair mRNA decay and cause toxicity. "In the future, there might be a combined therapeutic strategy where you also push it off Edc4 to fine-tune the effect,” Hallacli suggested.
Khurana believes α-synuclein’s membrane and P-body pathologies may reflect different avenues to disease vulnerability in patients. His lab is examining a potential physiological role for α-synuclein in P-bodies. If it has one, that could affect strategies that seek to suppress the protein via antisense oligonucleotides or immunotherapy (May 2018 conference news; Apr 2021 conference news; Apr 2022 conference news). “Understanding the functions of α-synuclein will be important to pinpointing undesirable on-target effects of such approaches,” Khurana wrote to Alzforum.—Madolyn Bowman Rogers
References
News Citations
- Excess α-Synuclein Sends Synapses Sputtering
- Synuclein Oligomers: Is EnSNAREing Synaptic Vesicles Their True Calling?
- Fatty Acid Greases the Wheels for α-Synuclein Multimers
- Lindquist Leaves Behind Parkinson’s Interactome as Her Parting Gift
- Parkinson’s GWAS—Genes Could Explain a Quarter of Late-Onset PD Risk
- Lack of C9ORF72 Protein Renders Neurons More Vulnerable to Degeneration
- Dysfunctional Lysosomes Cause Ferroptosis in Neurons
- At AAN, Sights Set on Antisense Therapies for Diseases of the Brain
- For α-Synuclein Immunotherapy, Is Going Later the Key?
- UB-312 Synuclein Vaccine Safe in Controls. Next Up: Parkinson's.
Paper Citations
- Corbet GA, Parker R. RNP Granule Formation: Lessons from P-Bodies and Stress Granules. Cold Spring Harb Symp Quant Biol. 2019;84:203-215. Epub 2020 Jun 1 PubMed.
- Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, International Parkinson's Disease Genomics Consortium, 23andMe Research Team, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, Graham RR. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet. 2017 Sep 11; PubMed.
Further Reading
News
- Parkinson’s GWAS—Genes Could Explain a Quarter of Late-Onset PD Risk
- With Age, Slower Protein Turnover May Predispose to Neurodegeneration
- Survey of Tau Partners Highlights Synaptic, Mitochondrial Roles
- TWAS x GWAS? Transcriptome Analysis Finds 11 Parkinson’s Genes
- UB-312 Synuclein Vaccine Safe in Controls. Next Up: Parkinson's.
- Yeast Screen Implicates PARK9 in Synuclein Toxicity
Primary Papers
- Hallacli E, Kayatekin C, Nazeen S, Wang XH, Sheinkopf Z, Sathyakumar S, Sarkar S, Jiang X, Dong X, Di Maio R, Wang W, Keeney MT, Felsky D, Sandoe J, Vahdatshoar A, Udeshi ND, Mani DR, Carr SA, Lindquist S, De Jager PL, Bartel DP, Myers CL, Greenamyre JT, Feany MB, Sunyaev SR, Chung CY, Khurana V. The Parkinson's disease protein alpha-synuclein is a modulator of processing bodies and mRNA stability. Cell. 2022 Jun 9;185(12):2035-2056.e33. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
U.K. Dementia Research Institute at University College London
In their study, Hallacli and colleagues reveal a completely new physiological function for α-syn, i.e., the regulation of mRNA degradation via direct modulation of Edc4, a protein important for P-body function. To show this, they use a variety of systems, from yeast to human patients, and unbiased screening methods complemented by hypothesis-driven mechanistic experiments.
While this function has not been reported previously, and it seems puzzling how it could have been overlooked, the evidence presented here is quite convincing, and the observations actually fit well in the current literature.
For me personally, the specificity of the P-body modulation to the N-terminus of α-syn and the successive mutual exclusivity of α-syn interaction with P-bodies and membranes is the most interesting part of this story. Our and other labs have found that increased interaction of α-syn with lipids seems to correlate better with disease state in human patient samples, and exerts stronger toxicity in cell and mouse models, than amyloid fibril formation in those same systems.
How aberrant membrane interaction causes toxicity has been somewhat unclear since it seemed to be exerted by physiologic α-syn and not pathological oligomers, but the assumption was that it just interferes with vesicle trafficking, based on work by Susan Lindquist in the early 2000s. The study here would imply that aberrant lipid binding does not necessarily cause dysfunction via the membrane, but by preventing proper α-syn-P-body interaction as shown here by the experiments in cell culture using the “3K” mutant of α-syn.
If the findings hold up to be disease-relevant, there will be a plethora of new exploratory studies into the outcome of impaired P-body modulation, starting a completely new field of α-syn biology with the potential to answer some longstanding questions in the field, e.g., brain region specificity or selective neuronal vulnerability based on the respective local RNA stabilization by synuclein dysfunction.
On the flip side, way more research in this direction is needed to evaluate the therapeutic potential of this finding, since the current study does not go deeper in that direction. Also unclear is how this relates to amyloid aggregation of α-syn, since the results in this regard are still early days. The correlation of Lewy bodies with mRNA accumulation in human tissue, and the experiments in cell culture with PFF seeding presented here, imply that α-syn aggregates lead to increased mRNA stability, but as said, more research is needed.
In any case, I think this paper will be a seminal publication in the field.
Yale University
I read this paper with great interest. It raises many exciting questions, both on the physiological function and pathological roles of α-synuclein, for instance, why only α-synuclein and not other family members bind P-bodies when the N-terminal region is pretty conserved? Also, what determines the switch between binding membranes versus P-bodies?
The authors nicely show that mRNA stability is altered in human neurons, but it remains to be seen if P-body modulation is contributing to these effects. The easiest way is to test this in SNCA knockout human neurons.
More remains to be done before we can begin to think about therapeutic implications.
Boston University School of Medicine
This article by Hallacli et al. from the Khurana lab opens up new avenues for the biology and pathophysiology of α-synuclein. The work shows that α-synuclein binds to P-body elements and promotes formation of micro-P-bodies in a dose-dependent manner, while pathological α-synuclein isoforms as well as aggregated α-synuclein can inhibit formation of macro-P-bodies. The authors do a good job of linking this biological pathway to Parkinson genetics and showing the biology in iPSC-differentiated neurons from subjects with varying dosages of α-synuclein, and showing that this modulates RNA catabolism.
In this context, this opens up fascinating new biology for α-synuclein. My group had looked at the relationship between α-synuclein and stress granules years ago (in collaboration with the late John Trojanowski), but never saw anything convincing. Hallacli’s work perhaps now explains why, but also shows an intriguing interaction with RNA metabolism paralleling that seen with RNA-binding proteins linked to ALS as well as for MAPT. Thus, this work extends the emerging link between RNA metabolism and neurodegenerative disease.
I echo the comments of Tim Bartels. His work had pushed a hypothesis that α-synuclein formed a complex that is associated with lipid membranes, but disruption of the complex leads to cytoplasmic α-synuclein, which could be toxic. They focused on brain and red blood cells and did not examine physiological pathways such as RNA metabolism, but I am struck by the way in which Bartels and Hallacli’s stories both posit an equilibrium between membrane-bound and cytoplasmic α-synuclein.
The stories differ in the biological outcomes examined. Bartels’ group, with Dennis Selkoe, focused mainly on aggregation, while Hallacli’s work focuses on a physiological outcome, which can impact on pathophysiology when α-synuclein aggregates are present. Nevertheless, both stories point to a biological equilibrium between membrane-bound and membrane-free α-synuclein, which therefore opens the way toward future studies examining the regulation of this equilibrium and these interactions.
Make a Comment
To make a comment you must login or register.