CONFERENCE COVERAGE SERIES
Alzheimer's Association International Conference 2020 (Virtual Meeting Only)
Amsterdam, Netherlands
27 – 30 July 2020
CONFERENCE COVERAGE SERIES
Amsterdam, Netherlands
27 – 30 July 2020
Suddenly, phospho-tau217 looks to be the most robust plasma biomarker for Alzheimer’s disease yet. That’s the general—and enthusiastic—consensus from two papers and several presentations at this year’s virtual Alzheimer’s Association International Conference, being held July 27 to 31.
In one study, the marker identified people with the highest likelihood of Alzheimer’s disease, as judged by neuropathology of plaques and tangles, with an AUC or 0.98, i.e., almost 100 percent accuracy. Postmortem neuropathology remains the gold standard for AD diagnosis. In differential diagnosis, plasma p-tau217 also distinguished AD from other neurodegenerative diseases, notably including tauopathies, with high accuracy, beating out other plasma markers including neurofilament light and p-tau181.
“I am tremendously excited about the potential for plasma p-tau217 to advance research, drug development, and care,” Eric Reiman, Banner Alzheimer’s Institute, Phoenix, told Alzforum. Reiman was a senior author on one of the studies, which was led by Oskar Hansson, Lund University, Sweden.
The new data comes from several labs using different cohorts and two types of test—ELISA and mass spectrometry. Hansson’s group, as well as researchers in Adam Boxer’s lab at the University of California, San Francisco, used an immunoassay developed by Jeffrey Dage at Eli Lilly, Indianapolis. Nicolas Barthélemy and colleagues in Randall Bateman’s lab at Washington University, St. Louis, used MS spectrometry to detect p-tau217 after concentrating plasma samples up to 800-fold. All told Alzforum they were extremely encouraged by their results and by the general agreement between their labs.
“From a clinical perspective, p-tau217 assays will be very useful in the future. I actually wish I could use it now, because it would make diagnosis so much easier,” said Boxer. Hansson expressed similar sentiments. “Given it is so accurate, a p-tau217 assay could easily be used to complement cognitive screening in memory clinicals that lack PET or CSF,” he said, adding, “It will be really useful in primary care to aid in diagnosis of people who never get to see a memory disorder specialist.”
The high accuracy of the plasma assay does not come as a complete surprise since, earlier this year, the Hansson and Bateman labs independently reported that p-tau217 in the cerebrospinal fluid outperformed other markers in diagnosing AD (Apr 2020 news). The question was whether p-tau217 in the plasma would do the same.
Using the Dage assay, Hansson’s group measured the marker in three different cohorts totaling 1,402 cases and controls. Co-first authors Sebastian Palmqvist and Shorena Janelidze tested 699 volunteers in BioFINDER-2, a Swedish prospective observational study; a neuropathology cohort comprising 81 AD cases and controls from the Arizona Study of Aging and Neurodegenerative Disorders/Brain and Body Donation Program at Banner Health, Phoenix; and 622 carriers and noncarriers of the presenilin 1 E280A Paisa mutation in Colombia. The data are reported in the July 28 JAMA online.
In the neuropathology cohort of 34 AD patients and 47 controls, plasma p-tau217 correlated tightly with neurofibrillary tangle density (see image below). It identified those with an intermediate-to-high likelihood of having had AD, as judged by CERAD criteria, with an AUC of 0.89. This compares with AUCs of 0.72 and 0.50 for plasma p-tau181 and neurofilament light (NfL), respectively.
The AUC, or area under the curve, reflects both the specificity and sensitivity of a test, with a value of 1.0 being perfection. When the authors considered only those brain samples that had a high likelihood of AD, i.e., with moderate to frequent plaques and Braak stage V/VI neurofibrillary tangles, the AUC for p-tau217 was 0.98.

Tangle Telltale. In a neuropathology series, plasma p-tau217 tightly correlated with the density of neurofibrillary tangles in people who had had Alzheimer’s, but not in controls.
Reiman was amazed at this level of accuracy. “Consider that this is only in those with amyloid plaques, and that this marker also distinguishes amyloid-related from primary tauopathies. What we have is a measure of amyloid-mediated tau pathophysiology, essentially an indication of plaques and tangles in one marker,” he said. To a large majority of Alzheimer’s researchers, amyloid-mediated tau pathophysiology defines Alzheimer’s disease.
The blood marker achieved similarly high accuracy when used to identify people who were still living with a clinical diagnosis of AD. In BioFINDER-2, p-tau217 distinguished 121 AD patients from 224 controls with an AUC of 0.98.
It differentiated those same AD patients from 99 other people with a different neurodegenerative disease with an AUC of 0.96. They included 12 people with vascular dementia, 45 with Parkinson’s disease or multiple system atrophy, 21 people with behavioral variant frontotemporal dementia (bvFTD) or primary progressive aphasia (PPA), and 21 with progressive supranuclear palsy (PSP) or corticobasal syndrome (CBS). Again, in this differential diagnosis, plasma p-tau217 outperformed plasma p-tau181, plasma NfL, and two imaging markers—cortical thickness and hippocampal volume (see image below).
Overall, the new marker was able to distinguish AD from any of these other diseases, or from people who had a mild cognitive impairment but whose Aβ levels were normal, with AUCs that were 0.92 or higher.

Differential Diagnosis. In BioFINDER-2, plasma p-tau217 accurately distinguishes Alzheimer’s from other neurodegenerative diseases, outperforming other plasma and imaging markers.
Elisabeth Thijssen, who worked in Boxer’s lab but is now at Amsterdam University Medical Centers, painted a similar picture of differential diagnosis using plasma donated by 210 people who sought care at the USCF Memory and Aging Center or enrolled in the Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) study. These samples comprised 37 healthy controls, 39 with a clinical diagnosis of AD, 35 with mild cognitive impairment, and 99 who had been diagnosed with some form of FTLD. The latter included 33 each with CBS or PSP, 23 with bvFTD, seven with non-fluent PPA, and three with semantic-variant PPA.
Using the Dage assay, Thijssen found that plasma p-tau217 separated AD from controls and from FTLD with AUCs of 0.92 and 0.86, respectively. This was slightly more accurate than p-tau181, which returned AUCs of 0.86 and 0.84. Among 76 volunteers who had had a flortaucipir PET scan, their plasma p-tau217 and p-tau181 values correlated with tracer uptake with AUCs of 0.96 and 0.91, respectively. “Both markers show good results, but I believe plasma p-tau217 is incrementally better,” said Boxer.
For his part, Barthélemy at WashU also found that the p-217 isoform outperformed p-181. He analyzed samples from two cohorts—a discovery cohort comprising 34 people who had enrolled in a stable-isotope-labeling kinetic study to measure Aβ production and clearance, and a validation cohort that included an additional 92 people enrolled in the same study. These data were published in the July 28 Journal of Experimental Medicine.
In the discovery cohort, both plasma p-tau217 and plasma p-tau181 correlated tightly with the respective concentrations of these same markers in the CSF—but only in people who had a positive amyloid PET scan. The p-217 and p-181 tau isoforms identified 15 Aβ-positive samples from 19 negatives with AUCs of 0.98 and 0.95, respectively. In the validation cohort, which had 50 Aβ-positive and 42 negative samples, the AUC for plasma p-tau217 fell slightly to 0.93, but here p-tau181 did poorly, with an AUC of 0.72.
Why does p-tau217 seem to be better than even p-tau181? For one, its dynamic range is luxuriously large. In BioFINDER-2, plasma p-tau217 was seven times higher in AD than in controls. That is fold. Not percent. Thijssen found it to be 5.7-fold higher in AD, while p-tau181 was 4.5-fold higher. These are expansive, newly comfortable ranges to work with for Alzheimer’s researchers, who are more used to wondering whether a 5 or 10 percent reduction in CSF tau in a therapeutic trial might be meaningful, or how robust an Aβ42/40 ratio change within a 15 percent range can be.
Barthélemy believes p-tau217 is the better marker because very little of it is present in control plasma. It is barely detectable in controls, whereas small amounts of p-tau181 are detectable. Herein lies the beauty of mass-spectrometry analysis. It detects both phosphorylated and unphosphorylated fragments, allowing Barthélemy and colleagues to calculate the occupancy of phospho groups at the various amino acids. What they found was telling.
In control CSF, about 2.6 percent of all tau fragments containing the 217 site were phosphorylated at that position. In control plasma, only 0.6 percent of the fragments were phosphorylated. Barthélemy thinks this is because the plasma contains some tau that originates from the body’s peripheral organs and is not phosphorylated at 217. In essence, in the blood, peripheral tau dilutes the tau that seeps out of the brain. For p-tau181, the scenario was slightly different. In the CSF, 13.5 percent was phosphorylated, while in the plasma 6.8 percent was. In essence, it seems that plasma contains about half as much p-tau181 as does CSF, but 4.3 times less p-tau217. “We drew the conclusion that some other source of tau in the plasma has a different phosphorylation status than does CNS tau,” said Barthélemy. “P-tau217 is highly specific to the CNS, explaining why plasma p-tau217 outperforms plasma p-tau181.”
Early Diagnostic Marker?
How soon in the trajectory of this decades-long disease can the increase in plasma p-tau217 be picked up? Clues to this question came from Hansson’s analysis of the Colombia cohort samples. Mean concentrations were 1.9 pg/mL in 257 members of the kindred who did not carry the Paisa mutation, 4.5 pg/mL in 259 cognitively unimpaired mutation carriers, and 16.8 pg/mL in 106 cognitively impaired carriers. In carriers, plasma p-tau217 rose over time, such that by 25 years of age, they had significantly higher levels than noncarriers. That’s about 20 years before the age of symptom onset (see image below). The data suggest that, like p-tau181, p-tau217 is detectable in the blood years before a clinical diagnosis and even before a positive tau PET test. Hansson told Alzforum that, in a follow-up manuscript, Janelidze describes plasma p-tau217 increasing before tau can be detected by PET, even in the entorhinal cortex, an early region for tangles. “Just as for CSF p-tau217, the plasma level increases first, showing it is a much earlier marker than PET.”

Early Marker. In people who carry the E280A presenilin 1 mutation, p-tau217 rises in the plasma about 25 years before symptom onset (left) and better distinguishes carriers from controls than does serum NfL (right).
Will p-tau217 become the first approved blood test for AD? Researchers agreed that more work remains to be done. Hansson said it is important to see if the test works in a primary-care setting and has begun to test this. A few months ago, he set up a prospective study, much like the IDEAS study for amyloid PET, to see if p-tau217 assays can improve routine AD diagnosis or change how physicians treat patients (Apr 2019 news). He had recruited about 60 volunteers in southern Sweden before COVID-19 slowed the study down. “This study represents an important step and we hope to pick this back up now,” said Hansson.
At the same time, he thinks the test soon can be implemented in clinical trials and even in retrospective testing of completed or ongoing studies. Banked samples could be tested quickly, including those from large epidemiological studies and clinical trials. Boxer said the plasma assay will be easier and more efficient to use than current PET- and CSF-based diagnostics. “It could lead to more creative and pragmatic trial approaches. The hope is it would help recruit more minority and underserved populations who maybe don’t have the resources to take part in studies that require more invasive tests,” he said.
Along the same line, Suzanne Schindler from WashU presented preliminary data from the Study to Evaluate Amyloid in Blood and Imaging Related to Dementia at AAIC. SEABIRD was designed to reflect the demographics of the St. Louis metropolitan area, improving racial, ethnic, educational, and socioeconomic representation in this research. Schindler reported that in blood samples from this population, plasma p-tau217 more accurately reflected a person’s amyloid status than did plasma p-tau181, suggesting that the 217 isoform performs well in this community setting.
Even so, it will take some time to get a plasma tau test ready for regulatory approval as a diagnostic. The assay needs to be validated in additional cohorts and between labs. Cutoffs need to be set, protocol standardized, certified reference materials to calibrate commercial assays need to be made. This kind of applied preapproval research takes at least a year. Additionally, the current immunoassay has a lower limit of detection of 0.48 pg/mL, which is not sensitive enough to measure the extremely low p-tau217 levels found in some control samples. “Ideally, we’d want to have some confidence that a test was negative because the amount of p-tau217 was really low and not because we couldn’t detect it,” said Barthélemy. With a lower limit of detection of 0.05 pg/mL, mass spec detected p-tau217 in all samples tested, but that required at least 4 mL of blood, which is orders of magnitudes more than routinely collected in certain studies, such as clinical trials.
It is possible that the sensitivity of the immunoassay might be improved. There is now a separate, new, single-molecule array assay for plasma p-tau181 developed by Kaj Blennow, Nicholas Ashton, and colleagues at the University of Gothenburg, Sweden, that seems more sensitive than the Dage assay for this form of tau (Apr 2020 news). In an upcoming paper, the Gothenburg group reports that plasma p-tau181, when measured up to eight years before a person’s death, predicts postmortem plaque pathology better than does a clinical diagnosis of AD, and that it distinguishes AD from other neurodegenerative disease with an AUC of 0.97. “We need some additional studies before we can say how much of a difference there is between these various p-tau biomarkers,” Blennow wrote to Alzforum. “Given that plasma p-tau181 performs quite well to identify AD and differentiate it from other diseases, I think differences will be relatively minor.”
Blennow further emphasized that the field needs to learn more about what phosphorylation of tau at different sites means in terms of AD pathophysiology. For example, are tau fragments of different sizes phosphorylated on different amino acids? If so, why? Do some have more than one phosphate and others none, and what would that mean?
Bateman thinks it’s important to better understand what the timing of phosphorylation implies about plaques and tangles. In the DIAN cohort, p-tau217 increases two decades before tau PET can detect neurofibrillary tangles. “Does this indicate that p-tau217 is a reaction to amyloid plaques that precedes tau pathology? Or is tau pathology beginning two decades before it is detectable by tau PET?” he asked (see comment below). Bateman also noted that p-tau217 decreases after symptom onset, while the tau PET signal and clinical dementia continue to increase. “This evidence strongly suggests that p-tau217 (and p-tau181) are not direct measures of tau pathology, but rather a reaction to amyloid plaques that later is associated to tau pathology,” he wrote. —Tom Fagan
No Available Further Reading
During a viral pandemic, everything to do with vaccination is au courant. At this week’s virtual Alzheimer’s Association International Conference, two reports drew attention by turning on its head the field’s emerging research trend suggesting that systemic infections could help trigger Alzheimer’s pathology in the brain. If that is true, the new studies asked, does warding off infection lower a person’s Alzheimer’s risk? Some initial data presented at AAIC hint that it might. Analyzing large population datasets, researchers found that vaccination against both influenza and pneumococci was associated with a lower risk of subsequent Alzheimer’s disease. Alas, the data are correlational, hence it is unclear if the shots themselves are protective, or if people who choose vaccination engage in other healthy behaviors that make them less likely to get Alzheimer’s.
“This research, while early, calls for further studies in large, diverse clinical trials to inform whether vaccinations as a public health strategy decrease our risk for developing dementia as we age,” Maria Carrillo of the Alzheimer’s Association said in a press release.
Even if the effect on AD risk is indirect, vaccinations may improve the overall health of Alzheimer’s patients. Another study found that people with AD are twice as likely to die after a serious infection as are cognitively healthy people, suggesting they have a heightened vulnerability and might benefit more from vaccination than same-age controls.
One of the most common vaccinations is the annual flu shot. About 45 percent of American adults got the vaccine in the 2018–19 flu season, according to the Centers for Disease Control and Prevention. Some previous studies have associated past flu vaccinations with a lower risk of dementia (Verreault et al., 2001; Liu et al., 2016).
To investigate further, Albert Amran, working in the lab of Paul Schulz at the University of Texas Health Science Center, Houston, assembled an ad hoc cohort from a medical records database. For the analysis presented at AAIC, they selected 9,066 participants over age 60, and matched Alzheimer’s patients with controls who had similar demographics. They also matched participants by medical history and access to care, to try to control for confounding effect due to better health habits. In this dataset, people who had received even one flu shot in years prior were 17 percent less likely to have AD than the unvaccinated group. Among the vaccinated subset, receiving yearly flu shots in the past dropped the odds of AD by another 13 percent. In addition, age at first vaccination mattered, with a one-year delay boosting the risk of later developing AD by 9 percent. Altogether, vaccinated people between the ages of 75 and 84 ran a 6 percent lower risk of developing Alzheimer’s over the next 16 years than their unvaccinated peers, the authors calculated.
“We hypothesize that vaccination may modulate neuroinflammation, resulting in a reduction of AD pathogenesis,” the authors wrote to Alzforum, noting that their final paper will include data on nearly 40,000 people.
In another study, flu vaccines by themselves were not protective, but pneumonia vaccines, or a combination of both, were. Svetlana Ukraintseva and colleagues at Duke University analyzed data from 5,146 participants in the NIH’s Cardiovascular Health Study. Half the cohort received one or more flu shots between the ages of 65 and 75, while 37 percent got vaccinated against pneumonia in the same time period. After the age of 75, 297 people, or about 6 percent of the cohort, developed AD.
People who got the pneumonia vaccine were 30 percent less likely to develop AD, after adjusting for sex, race, birth cohort, education, and smoking. Receiving more vaccinations seemed to bump up the protection, with a higher total number of flu and pneumonia vaccinations correlating with a 12 percent lower risk.
Intriguingly, the protection appeared stronger in people who did not carry the TOMM40 AD risk allele. This allele has been linked by some researchers to an earlier age of onset, although subsequent studies did not support the finding (Nov 2009 conference news; Jul 2010 conference news; Aug 2011 news). Among noncarriers of the proposed risk allele, receiving any pneumococcal vaccination was associated with 38 percent less risk of AD, while having a higher number of flu and pneumonia vaccinations dropped risk 15 percent in noncarriers.
Other researchers were divided over what the findings might mean. Rudolph Tanzi at Massachusetts General Hospital, Boston, speculated that systemic infections that spill into the brain could trigger β-amyloidosis, as has been demonstrated in model systems (May 2016 news; Jun 2018 news). Todd Golde at the University of Florida, Gainesville, said this mechanism could make sense given what is known about the immune system and AD, but cautioned that the current evidence is too weak to draw any conclusions about causation. “Inferring causation from [epidemiological] studies has often led the AD field down rabbit holes,” Golde wrote to Alzforum. Nonetheless, he noted that nearly everyone should receive influenza and pneumonia vaccinations anyway, due to their other beneficial health effects.
Another study indicated a need for this type of protection in elderly people with dementia. Janet Janbek at the Danish Dementia Research Centre in Rigshospitalet analyzed national health registry data on 1,496,436 people older than 65. She found that being admitted to a hospital with an infection tripled their death rate in general, but if the infected person also had dementia, their risk of dying was 6.5 times higher than that of non-demented controls.
However, other data cast doubt on the efficacy of vaccination in older populations. Lindsey Nakakogue at Pontifical Catholic University of Paraná in Londrina, Brazil, reported at AAIC that pneumococcal vaccination did not improve the odds of surviving pneumonia in a group of 510 people with dementia in their 80s. Slightly less than half received the vaccination, and both groups were equally likely to get pneumonia and to die from it.—Madolyn Bowman Rogers
The list of rare and common genetic variants that sway a person’s risk of Alzheimer’s disease just got substantially longer, as researchers expanded both the breadth and depth of genomic studies aimed at uncovering the etiology of the disease. At this week’s virtual Alzheimer’s Association International Conference, researchers described how harmonizing whole-exome-sequencing data from two massive consortiums in the United States and Europe unearthed rare variants that boost the risk of AD. In addition, geneticists extracted new information from existing genome-wide association studies by flexing their computational muscles of imputation, a feat that uncovered dozens of new AD risk variants, both common and rare. Other geneticists pulled out genetic variants that enhance cognitive resilience against AD. At this point, these lists look like little more than acronym salad, but scientists are quickly trying to find out what these genes are doing normally and in disease.
The AD field’s current genome-wide association studies (GWAS) have size on their side, but that’s not enough. They still lack the fine detail needed to capture the rarest variants. Sequencing studies are better suited to fish out these needles in a haystack, but most are limited by sample size. Researchers in Europe merged their sequencing data with data generated by researchers from the United States to address this issue. In the largest whole-exome sequencing study to hunt for AD variants to date, researchers led by Henne Holstege of the VU University in Amsterdam, Gaël Nicolas of Normandie University in Rouen, France, and Jean-Charles Lambert of the Institut Pasteur de Lille in France integrated exome sequences from the Alzheimer Disease European Sequencing (ADES) consortium with data from the American Alzheimer’s Disease Sequencing Project (ADSP), making for a total of 25,982 samples. The findings were posted in a manuscript in medRxiv on July 24, and presented at AAIC by co-first authors Holstege and Marc Hulsman, also at VU University Medical Center in Amsterdam.
Merging these massive datasets was quite a computational undertaking in itself, Holstege told Alzforum. In an effort led by Hulsman, the scientists combed through the data to weed out myriad batch effects from different sources of data. After extensive quality control, the dataset included exome sequences from 12,625 AD cases and 8,693 controls. While the harmonized dataset was generated and stored on the Dutch supercomputer facility in Amsterdam, the subsequent analyses were the result of a close collaboration by the Amsterdam group with the Lambert and Nicolas’ groups in France.
The researchers set out to discover rare genetic variants associated with AD risk. Specifically, they searched for individual variants that occurred in fewer than 1 percent of the samples, but lay within genes in which at least 10 people carried a variant. Out of a total of more than 7 million variants, the researchers detected more than 400,000 that were predicted to either wipe out expression of a gene or mess with its function. They separated them into four categories of badness, ranging from outright loss of function (LOF) to missense mutations with three degrees of predicted damage.
In the end, rare variants in 11 genes appeared linked to AD risk. Three of the 11—SORL1, TREM2, and ABCA7— had come up in previous studies. The others, in order of significance, were ATP8B4, ADAM10, ABCA1, ORC6, CBX3, PRSS3, B3GNT4, and SRC. Applying more stringent statistical techniques weakened some of these finds, but ATP8B4, along with the three known variant genes, remained significantly associated with AD risk. Variants in two of the suggestively associated genes—CBX3 and PRSS3—were less common in cases than controls, casting them as potential protective variants.
The average age at onset across all cases in the dataset was 73. For most of the variants tied to increased AD risk, carriers were younger at onset than noncarriers. Age at onset also decreased with increasing deleteriousness of a given variant; for example, symptoms emerged at an average of 60 years in carriers of LOF variants in SORL1, and not until 68 in carriers of milder missense mutations in the same gene. Variants in ATP8B4 did not appear to associate with age at onset.
A large proportion of the disease-associated signal from SORL1, ADAM10, ABCA1, ORC6, B3GNT4, and SRC came from singletons, that is, variants that were only detected in a single person. For TREM2, ABCA7, ATP8B4, CBX3, and PRSS3, the majority of the signal was derived from variants that were identified in multiple people, but were still very rare.
The findings presented in the medRxiv preprint need to be replicated in an independent set of sequence data from AD patients and non-demented individuals, Holstege noted. This is currently ongoing.
Most of the variants had some functional ties to AD pathophysiology, including APP processing and microglial function. The new risk gene ATP8B4, or ATPase Phospholipid Transporting 8B4, encodes a cation transport ATPase that ferries phospholipids across the cell membrane. In the brain, ATP8B4 is predominantly expressed in microglia, and rare variants in the gene have been associated with systemic sclerosis, an autoimmune disorder (Gao et al., 2016). The present study spotted 74 deleterious ATP8B4 variants in 767 people. Four percent of AD cases, compared with 2.5 percent of controls, carried one of the variants. Three missense variants drove the majority of the effect.
Holstege’s study was not the only one to present an ATP8B gene at AAIC. Another member of the family, ATP8B1, popped up in a genetic study of cognitive resilience. Timothy Hohman of Vanderbilt University, Nashville, Tennessee, reported results of a GWAS for variants associated with cognitive resilience to Alzheimer’s pathology. Essentially, Hohman looked for variants tied to superior cognitive performance in the face of a given amount of Aβ plaques. To assess resilience, Hohman integrated amyloid PET, postmortem neuropathology, and longitudinal cognitive performance data from more than 5,000 participants across four cohorts: Adult Changes in Thought (ACT), the Religious Orders Study and Memory and Aging Project (ROSMAP), the Alzheimer’s Disease Neuroimaging Initiative (ADNI), and the Anti-Amyloid Treatment in Asymptomatic Alzheimer’s (A4) study. For each participant, he charted Aβ burden against cognitive decline, developing a continuous variable of resilience. It ranged from those who performed worse than expected based on their pathological burden, to those who were sharper than expected. Every participant was genotyped.
Only one locus, by the ATP8B1 gene, associated with cognitive resilience at the genome-wide level. Three variants were detected near this gene. Further data will be needed to confirm that ATP8B1 is the gene causing the association, but Hohman said that variant carriers had reduced methylation of the gene, suggesting the variants may have increased its expression.
Hohman told Alzforum that the gene is highly homologous with ATP8B4 detected in the Holstege study, but its expression pattern is different. In the brain, ATP8B1 is expressed predominantly by neurons and by endothelial cells, which form the blood-brain barrier.
In his talk, Hohman focused on the liver, however, where ATP8B1 is known to play a role in regulating the homeostasis of bile acids. Variants in the gene have been tied to liver diseases. Interestingly, Hohman noted that data from ADNI indicate that the composition of bile acids change in AD. Hohman said future studies will need to unravel how ATP8B family genes influence AD risk and cognitive resilience. He noted that the ATP8B ATPases interact with ABC transporters, such as ABCA7, which are known to play a role in lipid metabolism and have been tied to AD risk.
Hohman also reported that the genetic architecture of cognitive resilience is distinct from that of Alzheimer’s disease. In other words, resilience is not simply the opposite of susceptibility. For example, Hohman reported that the genes associated with resilience in his study overlapped significantly with those tied to cognitive performance and education, and modestly with vascular and psychiatric phenotypes. Genetic pathways underlying AD risk were distinct from those tied to resilience.
Growing Larger and Denser, Monster GWASs Spew More Variants
Sequencing data may be the gold standard for pinpointing rare variants, but genome-wide association studies have the edge on size. They are massive, and growing larger still as consortiums pool their data.
Another way to get more variant bang per GWAS buck is to include more single-nucleotide polymorphisms in the analysis. To do this, geneticists have honed imputation methods. Essentially, they use haplotype reference catalogues to infer a genotype at one SNP position based on the genotype at another one nearby. Such catalogues have burgeoned in recent years, enabling the imputation of more SNPs than ever before, said Adam Naj of the University of Pennsylvania in his AAIC presentation. Naj used data from the Haplotype Reference Consortium, which includes nearly 65,000 haplotypes and 40 million SNPs, to impute data from the International Genomics of Alzheimer’s Disease Project (IGAP). Based on more than 25,000 AD cases and 40,000 controls, this GWAS had previously helped identify 25 risk loci (Mar 2019 news on Kunkle et al., 2019).
With deeper imputation, Naj pulled out 14 new common variants plus eight rare ones. Of the 14, only two—HBEGF and BZRAP1—reached genome-wide significance, while a dozen others tickled the significance threshold. They are EIF4G3, EPHA5, ELL, LOC102723838, MTSS2/1L, CLNK, LOC728084/LINC02458, SPPL2A, ANKRD33, LUZP2, GUCY2EP/TSKU, and MC4R. EPHA5 belongs to the ephrin receptor family, of which other members have been implicated in AD. SPPL2A is involved in innate immunity. MC4R encodes a melanocortin receptor that has been tied to obesity, and MC4R agonists reportedly boost neurogenesis and cognition in animal models of AD.
The eight new rare variants were in the genes, NOL10/ODC, LINC00052, LOC101927787, CCDC102B, NCK2, RORA, BSGALT6, and EPHA3, yet another ephrin receptor. Naj said that NCK2 reportedly interacts with presenilin. RORA encodes a nuclear receptor that regulates BDNF and insulin peptides in hippocampus. Finally, B4GALT6 plays a role in neural differentiation.
The imputed GWAS data need replication. Another consortium is already on it. At AAIC, Céline Bellenguez from Institut Pasteur de Lille in France reported preliminary results from a GWAS including data from the European Alzheimer’s Data Bank (EADB). When combined with data from seven smaller consortia, this GWAS analyzed more than 36,000 AD cases and 63,000 controls. Samples from some of the smaller consortia were also included in the IGAP GWAS; however, the majority of samples for IGAP came from the Alzheimer’s Disease Genetics Consortium, which were not included in the EADB GWAS. Bellenguez imputed this GWAS with the Trans-Omics for Precision Medicine (TOPMed) reference panel, comprising more than 125,000 haplotypes.
Bellenguez reported 44 loci tied to AD risk. Fourteen were new: COX7C-RASA1, RASGEF1C, UMAD1, CTSB, ZFPM2, TRIB1, C1S, TBX6, FAM157C, LILRA2, LILRB5-LILRB2, IDUA, GRN and—lo and behold—ATP8B4, the gene found in the WES study. LILRB genes are expressed in microglia and bind Aβ peptides. IDUA and GRN have been implicated in Parkinson’s disease and frontotemporal dementia, respectively.
Among the genome-wide significant hits, none popped up in both the EADB and IGAP imputed GWASs; but Naj noted that hits below the significance threshold may show some overlap. Plans are in the works to combine data from IGAP and EADB GWAS, which will further grow the sample. In addition, Bellenguez said they’ll add samples from UK Biobank soon (Apr 2018 news).—Jessica Shugart
Numerous studies have now found that amyloid PET scans can alter a person’s diagnosis and disease management—but do they reduce overall medical costs? According to data from the final phase of the IDEAS study, presented at the virtual Alzheimer’s Association International Conference this past week, the answer is no. Gil Rabinovici at the University of California, San Francisco, reported that patients who received a scan had about 5 percent fewer hospitalizations in the next year than did unscanned, matched Medicare participants. This small benefit missed the prespecified outcome of 10 percent, and leaves unclear whether insurers will consider the benefits of scanning to be worth the cost. Meanwhile, the researchers continue to dig through the dataset for additional insights, and IDEAS participants have joined additional studies.
“Our goal was to provide a resource to the field, beyond just addressing the issue of amyloid PET coverage,” Rabinovici told Alzforum. “We hope this will be a platform to pioneer the implementation of biomarkers and other research advances into the real world.”
The first phase of the IDEAS study found that physicians changed treatment plans for about two-thirds of the 11,409 people who underwent amyloid scans (Aug 2017 conference news; Nov 2018 conference news; Apr 2019 news). So far, so good. In the second phase of the study, however, the researchers compared hospitalizations and emergency-room visits of 12,684 IDEAS participants with those of the same number of matched controls from the Medicare database. In the year after an amyloid scan, IDEAS participants were 4.5 percent less likely to be hospitalized than controls were, but equally likely to visit an emergency room. The difference in hospitalizations was driven by the 4,379 participants with dementia, not the 8,305 with mild cognitive impairment.
Curiously, among the IDEAS participants, having a positive amyloid scan was associated with better health outcomes afterward than was having a negative scan. For both the MCI and dementia groups, people with positive scans were 22 percent less likely to be hospitalized in the next year than were those with negative scans. The amyloid-positive dementia group was also 15 percent less likely to visit an emergency room.
One possible explanation for this counterintuitive finding is that a positive scan and Alzheimer’s diagnosis led to an improved management plan that benefited patients’ health. Participants with a negative scan, on the other hand, continued without a diagnosis for their neurological issues, and thus their conditions were less well-managed and they were likelier to need hospitalization. That idea is consistent with the medical literature for other diseases, which links the presence of a clear diagnosis and multidisciplinary treatment plan to a decline in hospitalizations, Rabinovici noted.
However, an alternate explanation is that amyloid-negative participants simply have conditions that are more likely to require hospitalization. Rabinovici said their model tried to control for that by matching participants based on previous hospitalizations. The researchers are now analyzing the nature of the hospitalizations to determine if there are consistent health differences between amyloid-negative and -positive participants.
It is unclear if these data will convince the Centers for Medicare and Medicaid Services to extend coverage for amyloid PET scans (Jan 2013 news; Jul 2013 conference news). Rabinovici noted that CMS is expected to base their decision on the data from IDEAS and other studies to date. He believes the advent of accurate blood-based biomarkers might actually make PET scans more economical on a population level, by cutting down the number of people who would need one (Jul 2020 conference news). People could first be screened with a cheaper blood test, and then only those who tested positive would go on to receive PET scans or lumbar punctures for confirmation.
“I hope the totality of advances in biomarkers will be considered in coverage [decisions], and be part of an algorithm we’ll use in the future about how to deploy these biomarkers in clinical practice,” Rabinovici said. If aducanumab or another disease-modifying treatment is approved, the cost/benefit calculation would change (Jul 2020 news).
The researchers are digging further into the IDEAS data. “This is a treasure trove of data on over 18,000 people studied in real-world memory clinics,” Rabinovici said. He believes this dataset holds answers to many questions. For example, are claims-based diagnoses used in epidemiologic studies less accurate than case-report forms from dementia specialists? What conditions do amyloid-negative memory clinic patients have? By examining racial and ethnic groups in the IDEAS cohort, the researchers have found preliminary evidence of differences between whites and people of color. White participants were more likely to enter the study at the MCI stage, while blacks and Latinos were more likely to join at the dementia stage, suggesting a delayed detection of disease. At every disease stage, whites were more likely to be amyloid-positive than were blacks and Latinos, hinting that other conditions such as vascular cognitive impairment might be more prevalent in minority populations.
The next IDEAS study will address racial disparities by recruiting a diverse cohort. Whereas IDEAS participants were predominantly white, IDEAS 2 will include at least 2,000 blacks and 2,000 Latinos among the 7,000 planned participants. Outreach will be led by Consuelo Wilkins at Vanderbilt University in Nashville, Tennessee, and Peggye Dilworth-Anderson at the University of North Carolina, Chapel Hill. The study will also collect DNA and plasma samples and bank them in a biorepository for future research on predictive genetics and blood-based biomarkers. “There’s a need to understand MCI and dementia better in these communities that seem to be at higher risk of cognitive decline,” Rabinovici noted. The study is set to start this fall, and researchers are adjusting the protocols to adapt to increased use of telemedicine and heightened safety precautions during the coronavirus pandemic.
Meanwhile, the original IDEAS cohort has proved a fertile ground for recruitment to other studies. About 2,000 IDEAS participants contributed DNA to a study led by Andrew Saykin and Tatiana Foroud at Indiana University in Indianapolis. Another 1,000 joined the Brain Health Registry run by Michael Weiner at UCSF. Others are participating in a study of how amyloid PET scan results affect caregivers, while an add-on study will compare the accuracy of the plasma Aβ test developed by C2N Diagnostics with PET scans (Jul 2017 conference news; Aug 2019 conference news). “This unique cohort will allow us to address other research questions in the field and contribute to our knowledge about AD and related disorders,” Rabinovici said.—Madolyn Bowman Rogers
No Available Comments
At this year’s virtual Alzheimer’s Association International Conference, researchers presented an update to “Dementia prevention, intervention, and care,” the 2017 report commissioned by The Lancet. Published July 30, the new report identifies three additional modifiable risk factors for dementia, bringing the total to 12. Excessive alcohol consumption, air pollution, and traumatic brain injury cause 6 percent of all dementia cases, according to the international panel of experts led by Gill Livingston at University College London. In total, 40 percent of dementia can be prevented, the panel concludes, up from its 2017 estimate of 35 percent.
The panel revised slightly downward the risk that stems from low-quality education. Also new this year, the report addresses the risk COVID-19 poses to people who already have dementia.
The report’s 28 authors hail from Australia, Europe, India, Israel, Nigeria, and North America. They include clinicians, psychiatrists, epidemiologists, gerontologists, and public health experts. They reviewed and updated the work of the 2017 commission, which was convened to spotlight immediate opportunities for preventing dementia or improving care.
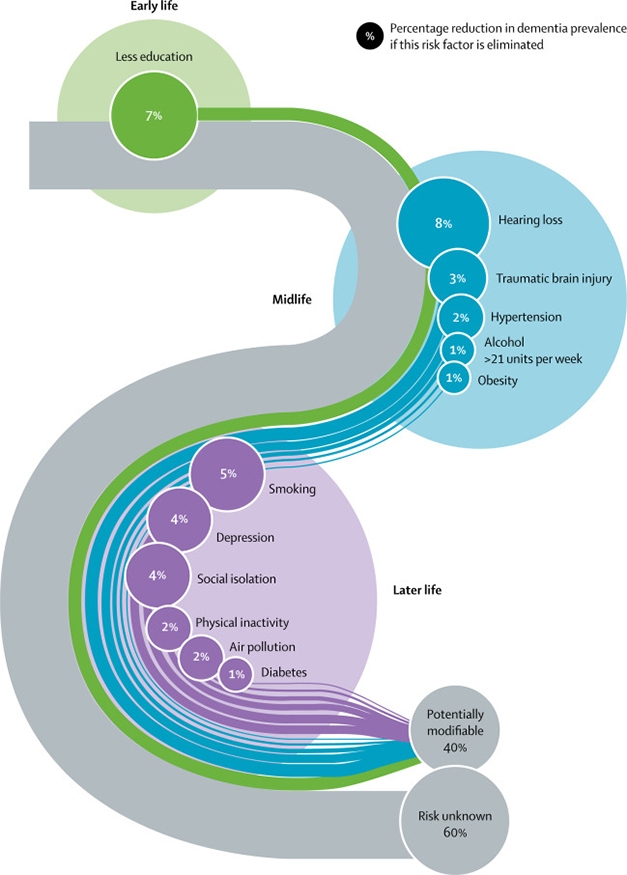
Life Course. This updated model stages 12 modifiable risk factors that together cause 40 percent of dementia cases. Traumatic brain injury, excess alcohol, and air pollution are the new additions. Numbers denote the fraction of the total prevalence attributed to each factor. [Courtesy of Livingston et al., Lancet 2020.]
The 2020 commission’s conclusions come from extensive literature review and meta analyses, including three new studies on alcohol consumption, four on traumatic brain injury, and one study of all-cause air pollution and all-cause dementia among more than 2 million people, average age 67, living in Canada.
According to the report, more than 21 units of alcohol per week is bad news for the brain. Alas, for anyone who still thinks—as scientists have said for years—that what is casually referred to as moderate drinking is good for you, consider this: The report defines a unit as 10 mL of pure alcohol, not a single alcoholic drink. In the U.S., a 12 oz beer at 5 percent alcohol contains 1.775 units, while a large glass of 12 percent wine contains about three units. Hence, two “drinks” per day increase a person’s risk of dementia. A recent French study of more than 31 million people who had been discharged from the hospital concluded that heavy drinking or alcohol dependence tripled a person’s risk for dementia (Feb 2018 news).
Traumatic brain injury, which can be as mild as a concussion or as severe as a skull fracture, increases a person’s risk for dementia by 80 percent, according to the report. TBIs typically come from car and bicycle accidents, sports injuries, military conflicts, or simple falls. TBI accounts for 3.4 percent of all dementia cases. Pollution accounts for 1.1 percent, but this may be higher in low-income communities. All types of air pollutant, including those from traffic and residential heating, have been associated with increased dementia incidence (May 2020 news).
In sum, the panel delivered eight messages for policy-makers and individuals, and recommended 14 strategies for reducing dementia risk (see box below).

Calling for an ambitious approach to dementia prevention, the scientists note inequalities that put certain groups at higher risk, and warn of a growing dementia incidence confronted by low- and middle-income countries.
Regarding COVID-19, the authors acknowledge that people with dementia are particularly vulnerable because of their age, co-morbidities, and difficulty keeping physical distance. They calculated that in nursing homes, people with dementia were 70 percent more likely to die if they contracted the virus than were residents without dementia. Even so, prevention and treatment strategies, such as isolating patients or transferring them to another location or hospital, can accelerate their decline. The commission recommends vigilant use of personal protective equipment and hygiene for staff, regular SARS-COV2 testing, and making oxygen available for patients who catch the virus but do not want to be transferred to a hospital.
The panel argues for a holistic approach to care, including management of neuropsychiatric symptoms. A holistic approach includes spotting and helping distressed caregivers to alleviate their anxiety, depression, and exhaustion. “I think the caregiving field is a pretty barren area of study, and the Lancet Commission report offers us a way forward,” commission panelist Eric Larson, Kaiser Permanente Washington Health Research Institute, Seattle, told Alzforum.—Tom Fagan
No Available Comments
No Available Further Reading
Having established that poor cardiovascular health in midlife increases the risk of cognitive decline and dementia as a person ages, scientists are now pushing to see if the same relationship might hold true even earlier in life. Indeed, several presentations at the Alzheimer’s Association International Conference, held virtually July 27 to 31, reported that cardiovascular risk factors present even at the beginning of adulthood might also affect the risk of developing Alzheimer’s disease. Researchers linked conditions such as hypertension, high cholesterol, obesity, and diabetes in young adults to smaller brain volumes, poor performance on cognitive tests, and a higher risk of AD decades later. Altogether, the findings highlight the importance of starting preventative lifestyle interventions as early as possible.
Previous studies have built a broad consensus among researchers that hypertension in one’s 40s or 50s boosts risk of dementia later in life (e.g., Whitmer et al., 2005; Aug 2017 news; Jun 2018 news). Cardiovascular disease in the 50s ups the likelihood of amyloid plaques after age 70, and obesity in the 50s seems to bring on dementia sooner (Apr 2017 news; Whitmer et al., 2005; Mar 2009 news; Sep 2015 news).
But how early in life do the processes underlying these risk associations start? This is difficult to study because it requires cohorts that record health data over decades. At AAIC, Kristen George, University of California, Davis, pushed the age when vulnerability can be detected back to the 30s. She analyzed MRI data from 220 participants in the Kaiser Healthy Aging and Diverse Life Experiences (KHANDLE) study. All were enrolled in the Kaiser Permanente health care system in northern California and were 65 or older and dementia-free. The cohort was racially diverse, with approximately equal numbers of whites, blacks, Latinos, and Asians. All had had a health checkup decades earlier, at an average age of 32, that measured blood pressure, cholesterol, and body-mass index. At that age, about a third of the cohort had hypertension, a third high cholesterol, and a quarter obesity.

Benefits now, benefits later. Former first lady Michelle Obama, shown harvesting vegetables with students in 2013, used her time in the White House to advocate for healthy eating and physical activity. Recent studies correlate obesity in the 20s and 30s with increased risk of later cognitive decline.
Participants who at that young age had had a BMI of 25 or greater had a smaller hippocampal volume on average in late life than did their peers without a history of obesity. A BMI of 25 or more is considered overweight, while 30 or higher is considered obese. Likewise, a young-adult blood pressure of 130/80 or greater, or total cholesterol of 200 or more, associated with a smaller cerebral volume after age 65.
This brain shrinkage had functional consequences. Across the cohort, smaller hippocampal volume was associated with worse episodic memory, as assessed by the Spanish and English Neuropsychological Assessment Scales (SENAS) cognitive composite. Participants took this test at an average age of 75. Similarly, smaller cerebral volume associated with worse working memory and executive function on the SENAS, George reported.
The risk to the brain from obesity may start even younger than that. Chillingly, a previous study linked high BMI to smaller brain volume even in children. Researchers led by Jennifer Laurent at the University of Vermont, Burlington, analyzed data from the ABCD study of 3,190 children at 21 sites across the U.S. All were 9 or 10 years old. The higher their BMI, the thinner their cortex was, particularly in prefrontal regions. This thinning correlated with worse performance on tests of executive function (Laurent et al., 2019).
The KHANDLE imaging sample was too small to determine if risk varied by race. However, the National Health and Nutrition Examination Survey (NHANES) 2015–2016 data previously found the highest prevalence of hypertension to be among blacks, while blacks and Latinos were more likely to be overweight or have diabetes than whites and Asians.
At AAIC, Rachel Peterson at UC Davis reported cognitive data from the Study of Healthy Aging in African Americans (STAR), which examined the effect of cardiovascular risk factors on late-life brain health specifically in this population. STAR enrolled 710 African Americans from the Kaiser Permanente healthcare system. All were 50 or older when recruited in 2018–2020. Importantly, whether they had had hypertension, cholesterol, obesity, or diabetes in young adulthood had been previously assessed, at around age 19 for 165 participants, age 25 for 435, and age 39 for 110.
The researchers assessed cognition in STAR participants at a mean age of 68, using the SENAS. Participants who had two or more of these risk factors at a young age performed worse on tests of executive function and semantic and episodic memory. In addition, there was a trend for diabetes in adolescence to associate with poor executive function late in life, and hypertension in early adulthood to associate with poor episodic memory.
Other presentations at AAIC reinforced these findings. Adina Zeki Al Hazzouri of Columbia University in New York analyzed data from 2,909 participants in the Cardiovascular Health Study and 2,195 from the Health, Aging, and Body Composition study. Both are diverse cohorts, with an overall mean age of 73. Women whose BMI topped 25 between the ages of 20 and 49 had nearly twice the risk of dementia as their leaner peers. In contrast, for women weighed in their 50s and 60s, BMI had no measurable effect on later dementia risk. For men, a high BMI at any point from ages 20 to 69 boosted dementia risk by 35 to 50 percent. For both sexes, a high BMI after age 70 associated with lower risk, as others have found (Fitzpatrick et al., 2009; Strand et al., 2013; Pedditzi et al., 2016; Singh-Manoux et al., 2018).
Similarly, Xiaoling Zhang at Boston University linked unhealthy cholesterol between the ages of 35 and 50 to Alzheimer’s risk decades later. Among 3,224 participants in the Framingham Heart Study Offspring cohort, those who had high levels of LDL, the “bad” cholesterol, and low levels of HDL, the “good” cholesterol, in this age range were more likely to have AD after age 60. Statin treatment at the earlier time point lessened the risk, but the relationship between early life cholesterol and late-life AD remained statistically significant.
Researchers don’t fully know how metabolic factors heighten AD risk. One possibility is inflammation. At AAIC, Kristine Yaffe at the University of California, San Francisco, linked systemic inflammation at young ages to late-life cognitive problems. She analyzed data from 1,920 participants in the Coronary Artery Risk Development in Young Adults study. Their mean age was 33. All had repeated measurements of plasma C-reactive protein, a clinical marker of systemic inflammation, over the course of two decades. Those whose C-reactive protein crept up over time, or was high throughout adult life, were twice as likely to perform poorly on tests of executive function and processing speed at age 55 as were those with consistently low C-reactive protein.
Intriguingly, a recent mouse study from Tony Wyss-Coray and colleagues at Stanford University found that activated B and T cells begin to accumulate in adipose tissue during middle age, suggesting a possible link between obesity and inflammation (Schaum et al., 2020). Obesity is also emerging as a major risk factor for a severe course of SARS-CoV2 infection, which, in turn, is marked by an excessive, dysregulated inflammatory response.—Madolyn Bowman Rogers
No Available Comments
Without the millions of thriving synapses that link them, neurons in the brain are useless. The crumbling of these tiny connective structures is widely viewed as the proximal step to cognitive impairment in Alzheimer’s disease. Yet, researchers struggle to define exactly how and when synapses start to malfunction along the trajectory of the disease in the human brain. At this year’s virtual Alzheimer’s Association International Conference (AAIC), researchers described new techniques, such as mass synaptometry and digital spatial profiling, that reveal how synaptic changes relate to the clinical manifestations of the disease.
They reported that some synapses in the Alzheimer’s brain are riddled with phospho-tau and markers of cellular stress, while neurons in the brains of resilient fellow citizens have managed to exclude pathological tau from the synaptic sanctum, fending off damaging neuroinflammation. Other researchers brought imaging and fluid biomarkers of synaptic integrity into the growing AD biomarker toolkit, with the hopes of more accurately predicting cognitive decline.
In his talk, Thomas Montine of Stanford University spelled out the problem: Even though synaptic degeneration is a central event in AD, scientists lack powerful methods to study large numbers of individual synapses in the human brain. They can use high-resolution techniques such as electron microscopy to see small numbers of synapses, or they can analyze the contents of millions of synapses in bulk via synaptosome preps. Montine and others use fluorescence-based flow cytometry to investigate what’s inside individual synapses. However, fluorescence-based techniques can only analyze a handful of proteins at once.
To overcome these limitations, Montine and colleagues developed “mass synaptometry” (Gajera et al., 2019). Adapted from CyTOF—a technique that simultaneously detects many proteins at the single-cell level—mass synaptometry merges time of flight (TOF) mass spectrometry with flow cytometry. Essentially, instead of tagging antibodies with fluorescent molecules, researchers label them with heavy metals, each with a distinct mass that can be detected by mass spec. In so doing, they can find dozens of proteins of interest within individual synaptosomes.
Montine showed results from an analysis of 39 synaptic proteins, including those specific for certain cell and synapse types, as well as AD- and PD-related ones. The researchers isolated synaptosomes from postmortem sections of the caudate putamens, hippocampi, and prefrontal cortices of seven cognitively normal controls, nine people who had died with AD, and seven with dementia with Lewy bodies (DLB). An analysis of about 500,000 synaptosomes per sample yielded some striking trends.
Compared with hippocampal synapses from controls or DLB, synapses from AD brain contained an overabundance of p-tau. In contrast, Aβ levels in synapses of the prefrontal cortex—a region littered with Aβ plaques in AD—were no different between the groups.
Using a statistical cluster analysis called CITRUS to identify AD-specific patterns of protein expression in hippocampal synapses, Montine defined three major AD-related synapse types in AD brains: One type had a reduction of VGLUT receptors; another had an increase in p-tau; and a third was chock-full of proteins that denote cell stress and injury, including APP, BIN1, activated Caspase 3, 3NT, and K48. Notably, these three clusters of synapses were distinct from each other, and were absent in people with LBD.
The findings demonstrate a diversity of synaptic changes that take place in the AD brain, which bulk methods would not have detected, Montine said, adding that his group is now deploying machine learning to understand this diversity in more samples.
Data by Jamie Walker of the University of Texas Health Science Center, San Antonio, also placed the synapse at the center of AD. Walker analyzed proteins in individual cells with a multiplexing approach. Rather than focusing on synapses, she used digital spatial profiling to measure 84 proteins within selected cells in fixed hippocampal tissue. Using antibodies conjugated to UV-photocleavable oligonucleotides, DSP allows researchers to measure proteins in precise spatial locations within a tissue. DSP is most widely used to type cancers.
Walker measured neuronal proteins in hippocampal sections from six people who had died with AD and eight people with significant AD pathology who had been cognitively normal at death. This latter group, considered “cognitively resilient,” represents more than a third of samples from postmortem studies, and researchers urgently want to know the mechanisms that drive their resilience.
First, Walker compared, within a given brain, the protein profiles of tangle-bearing neurons to those without tangles in them. Besides detecting multiple species of phospho-tau in the tangle-bearing neurons, Walker also found elevated levels of proteins involved in APP processing and Aβ degradation, including PSEN1, BACE1, ADAM10, neprilysin, and insulin-degrading enzyme (IDE). This would suggest a link between Aβ and tau deposition in the same cell, Walker said.
Curiously, tangle-bearing neurons from resilient brains had a smaller uptick in these proteins, including phospho-tau species, than did tangle-bearing neurons from AD brains. In fact, only one protein was more abundant in tangle-bearing neurons from resilient brains: the presynaptic marker synaptophysin. Elevation of this synaptic vesicle component in tangle-bearing neurons suggests that synapses in resilient brains may have functioned better in the face of tau pathology. Walker also surveyed proteins in the vicinity of tangle-bearing neurons. She found that more proteins involved in oxidative stress and neuroinflammation surrounded tangle-ridden neurons in AD brains than in resilient ones, whereas synaptic and axonal proteins predominated in resilient brains.
What’s inside synapses also emerged as a major determinant of cognitive resilience in findings presented by Teresa Gómez-Isla of Massachusetts General Hospital, Boston. Gómez-Isla scrutinized a large cohort of postmortem brain samples collected at five Alzheimer’s Disease Research Centers. Twenty-eight had died with dementia and a significant burden of AD neuropathology; 61 had had normal cognition, including 27 with little to no AD neuropathology (controls), 20 with a moderate burden of AD pathology, and 27 with a high burden. The latter two groups were considered intermediate or highly resilient, respectively.
Quantifying Aβ plaques and neurofibrillary tau tangles in the entorhinal cortex (EC), superior temporal sulcus (STS), and prefrontal cortex, the researchers found that people in the highly resilient group had a similar burden of both types of pathology as did people with dementia. Those in the intermediate resilient group had a lower pathological burden. In cell-culture assays, tau proteins extracted from the brains of highly resilient people seeded tau aggregation as efficiently as did tau extracted from AD dementia brains.
This ostensibly equal pathological burden had dramatically different consequences in the brains of resilient people versus those with dementia. While resilient brains had as many neurons in the EC and STS as controls, AD brains had lost roughly half of their neurons in these regions. What’s more, their remaining neurons were sorely lacking in both pre- and postsynaptic markers, suggesting profound synaptic loss. Neuronal projections in the hippocampi of AD brains were tortuous and curvy—a sign of dystrophic neurites—while those in highly resilient and control brains remained straight.
Next, the researchers isolated synaptosomes from the brain samples, and analyzed specific proteins via western blot. While synapses from resilient and AD dementia brains had similar amounts of Aβ peptides, those from the latter had at least twice as much phospho-tau, corroborating Montine’s mass-synaptometry data. This synaptic p-tau was accompanied by drastic astrocytic and microglial activation, as well as higher concentrations of proinflammatory cytokines in the entorhinal cortices of people with AD. Resilient brains had no signs of glial activation or inflammation.
The researchers repeated their main findings in the Religious Orders Study and Memory and Aging Project (ROSMAP) cohort, using brains from 25 controls, 25 resilient people, 25 with AD, and 25 who were considered “frail.” This last group had suffered from cognitive impairment before death, despite having no signs of Alzheimer’s or other neuropathology. Interestingly, frail people had levels of microglial activation on par with people who had had AD dementia, casting inflammation as a unifying factor across different types of cognitive impairment. Finally, the researchers reported that synaptic p-tau and glial activation strongly correlated with cognitive decline in those same brain donors before they passed away. Aβ plaque burden did not.
Displaying a tau-PET scan lighting up brain, Gómez-Isla cautioned trial designers that PET or CSF markers of AD pathology may be insufficient to foretell who will develop cognitive decline. Adding biomarkers of synaptic erosion and neuroinflammation might better predict who might develop symptoms, and when, she said.
Along those lines, researchers presented data at AAIC on both PET imaging and CSF biomarkers of synaptic loss. Adam Mecca of Yale University reported that compared with 19 controls, synaptic loss was readily detectable in 34 people with AD using 11C-UCB-J, a PET tracer that binds to the synaptic protein SV2A (Mecca et al., 2020). In 10 cognitively normal people and 10 with AD, Mecca reported that synaptic loss correlated markedly with tau deposition as measured by SV2A and tau tracers. Synaptic loss in the entorhinal cortex correlated with volume loss in the hippocampus in AD, suggesting tau pathology may have gummed up circuitry from the ERC to the hippocampus, leading to degeneration.
Emma Commans of Vrije University, Netherlands, correlated tau- and SV2A-PET in seven people with AD. She saw an odd dichotomy based on a person’s overall tau burden. In those with high tau, brain regions that had more tau had fewer synapses. This might seem to make sense, but consider this: In patients whose total tau burden was low, regions with the most tau had more synapses. Using magnetoencephalography to gauge synaptic activity, Coomans found that in people with high tau pathology and synaptic loss, brain-wave oscillations slowed from alpha into the delta frequency. The bulk of Cooman and Mecca’s findings were presented earlier this year at the Human Amyloid Imaging meeting (Feb 2020 conference news).
Researchers also explored relationships between CSF markers of synaptic loss and neuroinflammation along the AD spectrum. Andréa Benedet of McGill University in Montreal measured four CSF synaptic markers among 115 cognitively normal participants, 41 with MCI, and 31 with AD in that center’s TRIAD cohort. Neurogranin, GAP-43, and SNAP-25 levels correlated with clinical diagnosis, meaning levels were highest in the AD group. Synaptotagmin 1 was highest in MCI. SNAP-25 tracked most closely with clinical symptoms, and also with amyloid- and tau-PET in AD-related brain regions. In people with AD, CSF SNAP-25 correlated tightly with CSF p-tau-181,with the CSF markers of inflammation sTREM2 and YKL-40, as well as with [11C]PBR28 (TSPO-PET), a measure of microglial activation.
Joanna Pereira of the Karolinska Institute in Stockholm showed data on how CSF markers of synaptic and axonal degeneration changed with the stages of AD in the Swedish BIOFINDER cohort. Among 35 participants with evidence of Aβ, but not tau pathology, the SNAP-25, GAP-43, and neurogranin markers of synaptic damage were higher than in 37 controls. However, among 53 people with both Aβ and tau pathology, only neurofilament light (NfL), a marker of axonal damage, had increased further. Pereira believes this could mean that early on, amyloid damages synapses, leading to further increases in Aβ and worsening of synaptic damage. This synaptic damage instigates tau hyperphosphorylation and neurofibrillary tangles, leading to axonal degeneration and ultimately, cognitive impairment.
This sequence may seem at odds with Montine and Gómez-Isla’s findings, which pegged synaptic p-tau, not synaptic Aβ, as the marker of synaptic destruction. It remains unclear how the timeline of changes in CSF and PET markers of AD pathology and synaptic damage fits with the timeline of a shifting protein milieu inside synapses. One thing is clear: Synaptic damage is the defining prelude to cognitive decline.—Jessica Shugart
No Available Comments
No Available Further Reading
At this year’s virtual Alzheimer’s Association International Conference, researchers from Novartis and the Banner Alzheimer’s Institute presented highly anticipated follow-up data from the massive Generation 1 and 2 Phase 3 trials. These were halted when volunteers on the BACE inhibitor umibecestat began to develop subtle memory problems. Since then, volunteers and researchers both have been asking if that decline would be permanent. At AAIC, API investigators presented hot-off-the-press data to indicate that no, it wasn’t.
The trials, supported by Amgen and the NIH, tested whether umibecestat would prevent Alzheimer’s dementia in healthy carriers of the ApoE4 allele. One ApoE4 copy increases a person’s odds of getting the disease three- to fourfold; two copies, up to 15-fold. Dosing ended abruptly last summer when a preplanned interim analysis detected worsening cognition in people taking the BACE inhibitor (July 2019 news).
In her AAIC presentation, Ana Graf from Novartis in Basel, Switzerland, showed that several months after participants had taken their last dose, no differences remained between active and placebo groups. Eric Reiman from Banner Alzheimer’s Institute in Phoenix reported the same for brain volume lost during the trial.
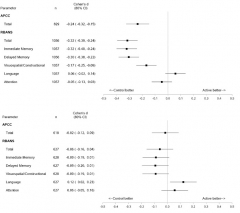
Cognition Restored. While taking umibecestat, volunteers performed worse on cognitive tests than did those on placebo, as seen in their last assessment on drug (top). Four months after they stopped, differences between the groups had all but disappeared (bottom). [Image courtesy API.]
Graf summed up data from the API Preclinical Composite Cognitive test and the RBANS. The APCC, designed to detect subtle cognitive change in healthy volunteers, was a primary outcome of the Generation trials. The RBANS, a commercial battery comprising immediate memory, delayed memory, attention, language, and visuospatial/constructional indices, was a secondary outcome; it was also used to assess safety during the trials. The RBANS was the principal measure that lead to the discontinuation.
To learn if the side effects were permanent, Graf and colleagues looked to volunteers who had taken 15 mg or 50 mg umibecestat daily for at least two months. These 1,056 participants had slipped on both the RBANS and the APCC, performing worse than those on placebo. Cohen’s d values of about 0.25 and -0.32 for the APCC and RBANS, respectively, indicated small to medium effect sizes. Poor immediate and delayed recall drove the RBANS deficit.
Graf noted that the difference between the drug and placebo groups was not wholly due to worsening on drug, but also to a greater proportion of people in the placebo group than on drug improving on the tests. Practice effects often lead to better cognitive scores as trials in asymptomatic or very mildly symptomatic participants progress. Practice effects have been a problem in the DIAN trials of solanezumab and gantenerumab, as well (Apr 2020 conference news).
What happened after volunteers stopped umibecestat? A median of four months later, the difference between placebo and drug groups had all but vanished, with Cohen’s d values of -0.02 for the APCC and -0.06 for the RBANS. The reversal occurred across all subgroups, namely people who were amyloid-positive or -negative at baseline and in both ApoE4 homozygotes and heterozygotes. The amyloid-positive homozygotes had declined most on drug, with Cohen’s d values of -0.55 and -0.42 for the APCC and RBANS, respectively, but following washout these differences had fallen to +0.01 for the APCC, and -0.12 for the RBANS. Graf concluded that there was no long-term impact on disease progression.
Reiman reached a similar conclusion based on MRI analysis. Pooled data from both trials indicated more loss of hippocampal and whole-brain volume in treatment groups by week 26, irrespective of dose. Cohen’s d values of -0.45 and -0.31 for whole brain and hippocampus, respectively, indicated medium effect sizes. The effect sizes were about the same at week 52, suggesting the volume loss was not progressive, said Reiman. Neither did it correlate with worsening total RBANS scores or with immediate or delayed recall.
Among 256 volunteers who had a follow-up MRI within two months of washout, their volume loss appeared at least partially restored. Cohen’s d values for drug/placebo comparisons of whole-brain and hippocampal volume had improved to -0.31 and -0.16, respectively. The difference in hippocampal volumes between drug and placebo groups was no longer significant.
Listeners wanted to know what caused the cognitive loss. Graf suspects umibecestat might have prevented BACE from cleaving substrates other than amyloid precursor protein, such as CHL1 or seizure protein 6 (Dec 2013 conference news; Oct 2016 conference news). She said that though Novartis has insufficient post-baseline CSF or PET data from Generation for further analysis, it is working on proteomics of CSF from previous three-month studies in healthy elderly volunteers to see if other BACE substrates might be affected and how that might relate to amyloid.
Others wondered how to interpret the loss of brain volume. Reiman believes it may reflect changes in the amount of brain fluid due to clearance of Aβ, an explanation proffered for other anti-Aβ therapies (Jul 2004 conference news; Dec 2017 news). In support of this, volunteers who were found to have elevated brain amyloid based on PET or CSF analysis had more brain shrinkage. “In general we are encouraged that we’ve seen only mild cognitive worsening, brain-volume changes that are not related to cognitive changes, and that all changes appear to reverse, or at least in case of MRI, begin to reverse,” said Reiman.
Umibecestat: Dead and Gone?
Graf told Alzforum that neither Novartis nor Amgen have plans for further development. Others believe BACE inhibitors may still prove useful. “We’ve got to think of the possibility that the drug deserves consideration for further use, as we begin to weigh the risks and benefits in future studies,” Reiman said.
Stefan Lichtenthaler, Technical University of Munich, agrees. “BACE inhibitors are promising, but need to be used at a lower dose (maximum 50 percent Aβ inhibition) and need to be given as a preventive drug and not for treatment of individuals already diagnosed with AD,” he wrote to Alzforum. He thinks 50 percent inhibition should be the max because mice missing one copy of the BACE1 gene appear normal.
Colin Masters, University of Melbourne, Australia, would dial the dosing down even further. Based on the rate of Aβ accumulation over a 20-year period, he believes 10 percent inhibition would be sufficient. He thinks the cognitive/psychiatric adverse events in Generation are due to umibecestat inhibiting the normal function of APP, which is needed for synaptic activity, learning, and memory. “The best strategy going forward, in my view, will be to use an antibody to lower the Aβ load to normal (might take a year or two) and then use a low-maintenance dose of a BACE inhibitor to keep it down,” he wrote to Alzforum.
If a BACE inhibitor is deployed in a future prevention trial, including a primary prevention trial in persons at genetic risk who do not yet have appreciable amyloid plaque deposition, then it will be important to consider ways to conduct the trial with adequate statistical power despite the modest worsening and very early preclinical stage, for example assessing persons with biomarker endpoints and/or with cognitive endpoints after temporary discontinuation of study drug,” Reiman wrote to Alzforum. “It will also be important to consider appropriate risk mitigation strategies.”—Tom Fagan
No Available Comments
No Available Further Reading
This year’s virtual Alzheimer’s Association International Conference featured no new efficacy data from large clinical trials, but there were some snippets of news from early phase studies. CT1812 by Cognition Therapeutics (CogRx) seemed to knock Aβ off its putative receptors in people with Alzheimer’s disease and reverse changes to the AD CSF proteome, while Alector’s passive immunotherapy against sortilin boosted progranulin levels in frontotemporal dementia patients. Boehringer Ingelheim outlined Phase 2 data that led it to discontinue its glycine transporter inhibitor for AD last February.
In preparation for an upcoming efficacy trial, researchers from CogRx chose proteomics to tease out effects of their drug candidate CT1812. Research indicates this antagonist of the sigma2 receptor displaces Aβ oligomers from prion receptors on the cell surface, reducing Aβ toxicity.
At AAIC, Susan Catalano from the Pittsburgh-based company presented data from the first study of 19 people with mild to moderate AD, who were treated for 28 days, as well as from the first 24 patients who completed a subsequent six-month Phase 2 trial. The latter, called SHINE, assesses safety, cognition, and biomarker measures of synaptic damage in 62 people with mild to moderate AD. Each received daily placebo, 100 mg, or 300 mg CT1812. Cognition as a primary endpoint was assessed with the ADAS-Cog 11 at baseline, then again at days 40, 100, and 185 of treatment. Three other trials of this drug are in progress.
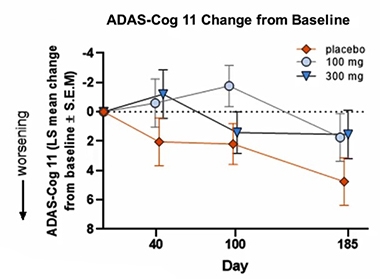
Separation? Among the first 24 patients tested in the small SHINE trial, those on drug appeared to fare better than those on placebo. [Image courtesy Susan Catalano, CogRx.]
For SHINE, Catalano reported a trend toward improvement on the ADAS-Cog11 in the drug group compared with placebo at the end of the six months of treatment. Other cognitive measures, including the MMSE, ADASCog13, and other dementia indicators such as attention and executive function all showed a trend toward improvement, she said.
Biomarker data from the 28-day trial of 19 people suggested the drug might be engaging its target as proposed. As measured by native western blots, Aβ oligomers in the CSF dropped by half from baseline in people on placebo, while they increased slightly in patients taking 90, 280, or 560 mg of CT1812 daily. Catalano showed only the percentage change for Aβ oligomers and only pooled data from the treatment groups because the number of patients in each was so low, she said. CSF levels of Aβ monomers stayed unchanged, as did SNAP-25 and neurofilament light. However, neurogranin and synaptotagmin-1 levels, as measured by ELISA, fell slightly, which might indicate fewer synapses are degenerating (Dec 2017 news).
To get an idea of what might be going on in neurons, the scientists used CSF proteomics to ask what changed in response to the drug. First, they looked at about 500 CSF proteins that previously had been implicated in synaptic biology (Lleó et al., 2019). Twenty-five of these, including proteins involved in synaptic structure and function, either rose or fell in the drug groups compared with placebo, suggesting something was happening to synapses.
Next, the researchers looked to see if CT1812 reversed changes seen in AD CSF relative to healthy controls (Higginbotham et al., 2019). In other words, if more of a given protein was found in AD CSF than in control CSF, would CT1812 squelch that difference or even reverse it? Again, about 20 proteins were shifted by CT1812, such that if they had been elevated in AD, they were suppressed in the drug arm, and if they had been suppressed in AD, they increased in the drug arm.
Lastly, Catalano reported that CT1812 decreased phosphorylation of tau at dozens of sites and increased it at six. Most of these sites are phosphorylated by enzymes other than GSK3β, though some GSK3β sites, including T205, S262, and T217, were less phosphorylated in the drug group. T181 seemed unaffected. Recent evidence suggests that T217 and T181 are phosphorylated very early in the disease trajectory in response to Aβ toxicity (Jul 2020 conference news).
What does this mean? It remains to be seen. Catalano said that the drug reverses 6 to 7 percent of the protein changes seen in AD. “We don’t yet know what it means to move 6 percent of those proteins after a month on drug,” she said. She noted it will be important to track these changes in future trials and also figure out if any of these markers correlate with cognitive scores. “In previous trials [of other drugs], we have seen changes in every single biomarker but no effect on cognition,” she said. CogRx is planning an efficacy study in 540 volunteers, sponsored by the NIA and to be run by the Alzheimer’s Clinical Trial Consortium.
A Boost for Progranulin
AL001 is an anti-sortilin antibody currently in Phase 1 trials for treating frontotemporal dementia in people who have progranulin haploinsufficiency. It, too, is showing promise based on biomarker readouts presented at AAIC.
The rationale with AL001 is that blocking sortilin-mediated endocytosis will extend the half-life of progranulin, improve lysosomal function, and slow or reverse clinical decline. In his presentation, Robert Paul, Alector, South San Francisco, reported results indicating that 12 days after a single dose of the antibody, progranulin in CSF had risen in six asymptomatic GRN mutation carriers. In eight symptomatic carriers, one dose of AL001 every two weeks for six weeks doubled CSF progranulin.
The treatment also reduced CSF levels of osteopontin and chitotriosidase, markers of inflammation and gliosis, respectively, and increased cathepsin B, an indicator of lysosomal function. CSF NfL, a marker of axonal damage, also trended down. Interim analysis of 10 symptomatic GRN mutation carriers enrolled in a Phase 2 trial suggested the antibody was well tolerated, Paul said. It restored plasma progranulin to normal levels beginning about 10 days after treatment began.
Lastly, in the didn’t-pass-muster category, Glen Wunderlich from Boehringer Ingelheim, Burlington, Ontario, reported that the glycine transporter BI 425809 inhibitor did not improve cognition in a 12-week Phase 2 trial. The company had announced this past February that it would stop development of this drug for AD, though a trial is still ongoing for schizophrenia.—Tom Fagan
No Available Comments
No Available Further Reading
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.