CONFERENCE COVERAGE SERIES
Society for Neuroscience Annual Meeting 2012
New Orleans, LA, U.S.A.
13 – 17 October 2012
CONFERENCE COVERAGE SERIES
New Orleans, LA, U.S.A.
13 – 17 October 2012
A National Institutes of Health initiative seeks to map the connections of the healthy human brain and provide a huge reference database freely available to researchers worldwide. The five-year “Human Connectome Project” began in autumn 2010, and its leaders provided a progress update in a symposium at the Society for Neuroscience 2012 annual meeting, held 13-17 October in New Orleans, Louisiana. James Bjork at the National Institute on Drug Abuse, Bethesda, Maryland, said the goal is to create a “gold-standard dataset” of brain wiring that “will have an immense impact on our understanding of disease states.”
To achieve this, NIH funded two parallel consortia under its Blueprint for Neuroscience Research, which tackles large projects beyond the scope of a single institution (see also NIH press release). Researchers at Washington University, St. Louis, Missouri, and the University of Minnesota, Twin Cities, lead the “WU-Minn” consortium. This project spreads over 10 institutions and costs $30 million. It will map the connectomes of 1,200 healthy young adults to high resolution and relate this structural data to functional, behavioral, and genetic information gathered on the same volunteers. The second consortium, based at Massachusetts General Hospital and the University of California, Los Angeles, focuses on building a state-of-the-art magnetic resonance imaging (MRI) scanner with “exquisite sensitivity” for imaging axons, said David Van Essen at WashU. All data from the two consortia will be made freely available through the Internet as it is generated. “We hope it will be a game-changer” for neuroscience research, Van Essen said.
This project will fill a gap in neuroscience, presenters said. Scientists understand less about how structure relates to function in the brain than they do for any other organ, said Van Wedeen at Massachusetts General Hospital, Boston. Connections between neurons define neuroanatomy, and these thin fiber tracts show up poorly with most imaging technologies. To visualize axons, researchers use a variant of MRI called diffusion tensor imaging, which maps the flow of water through tissues. As fluid moves along versus across axons, the technique allows scientists to see these microscopic projections. A recent advance, diffusion spectrum imaging (DSI), distinguishes intersecting tracts (see Wedeen et al., 2008). Crossing fibers cause a problem for dissecting brain structure, because multiple pathways can occupy the same location, Wedeen said. With these new techniques, “We can see things that cannot be seen with any other method,” Wedeen told the SfN audience. That includes visualizing the three-dimensional fiber architecture of the entire brain, he said (see video below).
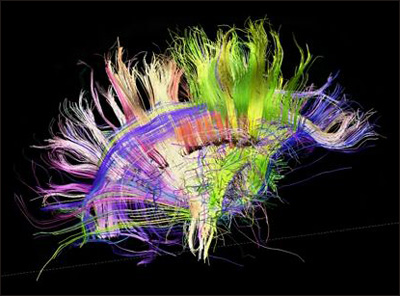
Brain wiring in a healthy adult, as seen by diffusion spectrum MRI. Source: Van Wedeen, M.D., Martinos Center and Department of Radiology, Massachusetts General Hospital and Harvard University Medical School
While maps of young adult brains will be informative, several audience members expressed interest in seeing a developmental time course of brain connectivity. At the moment, looking at younger brains or healthy older brains is beyond the scope of the project, said Story Landis, who directs the National Institute for Neurological Disorders and Stroke. If the community embraces the current database, however, future projects to look at connectivity in other populations might get funded, she added. Since equipment and infrastructure are now in place, such add-on projects would have a smaller price tag, Landis pointed out. Many researchers in the Alzheimer’s field have shown that brain connectivity in key regions decreases as the disease advances, making this a hot topic for AD research (see ARF related news story; ARF news story; and ARF news story).
"This project will be an important step in developing connectivity methods that can be used in AD research. The methods have great potential, and a lot of work is underway to enable optimal measurement stability for time series data," Keith Johnson at the Massachusetts Alzheimer's Disease Research Center, Boston, wrote to Alzforum. Other researchers agree. "The lessons we learn from the project could be easily translated to aging and AD. An understanding of what normal connections are will allow us to determine pathological changes," Beau Ances at WashU wrote to Alzforum. Neither researcher is involved in the project.
Van Essen gave an update on the WU-Minn consortium project, which he co-directs together with Kamil Ugurbil at the University of Minnesota. Phase 1, now complete, optimized data collection methods. In Phase 2, which began in August 2012, researchers collect data from volunteers. The 1,200 participants range from 22 to 35 years old and comprise 300 twin pairs (both identical and fraternal) and their siblings. The researchers chose this design in order to look at the heritability of brain circuitry patterns, Van Essen noted. So far, the consortium has scanned 12 participants, and posted a “starter kit” of data. They plan to have 80 participants scanned by the end of November, and will continue with quarterly data releases, the next one in February 2013, Van Essen said.
This project exemplifies “big data,” as it will generate about one petabyte, i.e., one quadrillion or 1,000,000,000,000,000 bytes of data, about 2,000 times the size of a typical desktop hard drive. During two days of testing, participants undergo a battery of imaging scans, including resting-state functional MRI (fMRI), task fMRI, diffusion MRI, and in some participants, magnetoencephalography and electroencephalography to look at brain electrical function on a millisecond time scale. During task fMRI, the volunteers perform seven exercises that engage different brain areas. These include tests of working memory, language, social cognition, and relational reasoning. Other tasks map the motor system, engage the reward system through a gambling paradigm, and evaluate recognition of facial emotions. In addition, the researchers collect demographic and behavioral data, and administer various tests of cognition and personality, including 19 tests from the NIH toolbox, a freely available set of cognitive, motor, sensory, and neuropsychological tests. In the fifth year of the project, the researchers will genotype participants using either single-nucleotide polymorphisms or whole-exome sequencing, depending on available funds, Van Essen said.
To use the data, other researchers can download the freely available Connectome Workbench platform. It allows visualization of the results in three dimensions and manipulation of the representation via toolbars. The Human Connectome data will link to other Internet resources such as the Allen Human Brain Atlas, which records patterns of gene expression in the brain. Gene expression patterns correlate well with functional connectivity, Van Essen noted.
The high-resolution connectome data generated by WU-Minn could have many uses, said Heidi Johansen-Berg at the University of Oxford, U.K., in her talk. For example, researchers might identify boundaries between brain regions by looking for changes in connectivity patterns. Using task fMRI data, researchers can ask questions such as, What brain networks co-vary with working memory performance? The findings will identify anatomical pathways relevant to task performance. For example, good two-handed coordination correlates with strong connectivity in a small cluster in the corpus callosum, Johansen-Berg said.
Presenters also touted the technical imaging improvements achieved by the WU-Minn consortium. Typical MRI images have a resolution of 2 or 3 millimeters, Johansen-Berg said. By contrast, the Human Connectome Project manages a resolution of 1.25 mm on its machines with 3 Tesla magnetic fields. Its scanners have a gradient strength of 100 mT/m, compared to the standard 40 mT/m, creating images with a higher signal-to-noise ratio and more clarity. All participants will be scanned on 3 T machines, with a subset also getting scans at 7 T and even on a 10.5 T machine under development, noted Ugurbil. These machines will provide higher resolution and contrast, and might allow researchers to see functional connectivity among fine structures such as cortical columns, Ugurbil suggested. Because diffusion MRI data contain some artifacts, researchers will validate the data against traditional tract-tracing experiments and correct images accordingly, Johansen-Berg noted.
The MGH/UCLA consortium hopes to generate even higher-resolution images. Wedeen, who co-leads the project, told the audience that a new Connectome scanner has a gradient strength of 300 mT/m, seven times that of conventional MRIs. Therefore, the scanner can acquire images in fewer milliseconds, providing a sharper signal because water has less time to wander. It will also reduce the scan times for participants, Wedeen said.
Wedeen shared some insights gleaned from DSI mapping to date. While neuroanatomy often looks confusing because of the crossing of multiple fiber pathways, the imaging resolved this into geometric order, showing that cortical fibers form smooth sheets that tend to travel in one of three perpendicular directions, forming a three-dimensional grid, Wedeen said (see Wedeen et al., 2012). These smooth sheets may reflect concentration gradients from embryological development, he speculated. Scientists have known for a long time that the cortex starts out as a flat sheet and crumples up during development into the deep convolutions of the adult brain. High-resolution microscopy also reveals that fibers typically make 90 degree turns rather than smooth curves when they need to change direction. This finding illustrates the power of the new machine to provide a more detailed anatomical understanding of the brain. Existing diffusion imaging methods cannot visualize these microscopic turns, Wedeen noted. Wedeen said he is impressed by the fixed patterns revealed by these scans. “The architecture of the brain is tremendously precise. This should be a huge asset for mapping and looking for changes in disease states,” he said.—Madolyn Bowman Rogers.
Polymorphisms in the gene for leucine-rich repeat kinase 2 (LRRK2) confer risk for both familial and sporadic forms of Parkinson’s disease (PD). LRRK2 substrates have been hard to pin down, and the kinase's role in pathogenesis remains unclear. At this year’s Society for Neuroscience annual meeting, held 13-17 October in New Orleans, Louisiana, several groups proposed myriad functions for LRRK2. If all those functions prove true, the kinase is one busy protein.
"We are at this early stage of research where we are just getting a handle on what LRRK2 is doing,” said Darren Moore of the Swiss Federal Institute of Technology in Lausanne (EPFL). “After we do that, we can work out which pathway is pathologically the most relevant."
Rachel Bailey, from Jada Lewis’ lab at the University of Florida, Gainesville, put forth the idea that LRRK2 phosphorylates tau. Hyperphosphorylated and aggregated forms of the microtubule-binding protein tend to afflict a subset of PD patients who have LRRK2 mutations. One common LRRK2 variant, with a glycine-to-serine substitution at position 2019 (G2019S), boosts kinase activity. Could that extra oomph be responsible for the increased tau seen in PD?
The researchers incubated LRRK2 with recombinant human tau in vitro and subsequently detected multiple phosphorylated sites on tau by mass spectrometry. The researchers then created a mouse model expressing both human wild-type LRRK2 and human mutant tau by crossing the BAC-LRRK2 mouse (Melrose et al., 2007) with the rTg4510 strain (Santacruz et al., 2005). Mice expressing both human genes accumulated more tau in their brains than did the tau transgenics, demonstrating that LRRK2 enhances tau pathology. Bailey presented no data on how these changes alter tau neurotoxicity, pathology, or behavior, but plans to address those points in future research.
A different presentation tied LRRK2 to a protein involved in Wnt signaling. Wnt is crucial in signal transduction, synaptic plasticity, and is increasingly implicated in neurodegenerative disease, said Daniel Berwick and Kirsten Harvey from the University College London School of Pharmacy, U.K. Previous results from Harvey’s lab pointed to LRRK2’s interaction with the disheveled family of phosphoproteins, important regulators of Wnt signaling (see Sancho et al., 2009). Berwick presented new yeast-two-hybrid screen, co-immunoprecipitation, and confocal microscopy data suggesting that LRRK2 directly interacts with another Wnt signaling component, i.e., low-density lipoprotein receptor-related protein 6 (LRP6). This transmembrane protein binds a cytoplasmic complex that destroys β-catenin while Wnt pathways are dormant.
Under basal conditions, LRRK2 resides in the cytoplasmic β-catenin destruction complex. Once the Wnt pathway is stimulated, however, LRRK2 binds to LRP6 in the membrane, and the two, together with the destruction complex, are taken up by endocytosis. With destruction complex components tied up in endosomes, β-catenin accumulates in the cytoplasm and eventually translocates into the nucleus, where it affects gene transcription.
Together, these observations suggest that LRRK2 bridges membrane and cytosolic components of Wnt signaling, said Berwick. Both a LRRK2 inhibitor and PD-associated LRRK2 mutations weakened the interaction with LRP6 and reduced Wnt signaling. Since those pathways are crucial for basic neuronal function and neurogenesis, their deregulation could lead to neurodegeneration, he and Harvey suggested in a related paper (see Berwick and Harvey, 2012). However, some scientists believe overactivation of the kinase contributes to pathology and are pursuing LRRK2 inhibitors as potential therapeutics. According to Berwick and Harvey’s data, inhibitors aimed at reducing LRRK2 activity may not be therapeutic if they also deregulate Wnt signaling, Berwick said.
Alexandra Beilina from Mark Cookson’s lab at the National Institutes of Health in Bethesda, Maryland, favors yet a different LRRK2 interactor. By protein microarray and co-immunoprecipitation, these researchers found that LRRK2 binds BCL2-associated athanogene 5 (BAG5) in both mammalian cells and in mouse brain. This protein acts as a co-chaperone for heat shock proteins, which are involved in protein folding activities in the cell, and has previously been implicated in Parkinson's (see ARF related news story). It is not clear what happens when LRRK2 binds BAG5. In Beilina’s hands, the interaction did not depend on kinase activity and LRRK2 did not phosphorylate the co-chaperone. Both proteins independently reduced neurite length when expressed in primary cortical neurons. BAG5 might act downstream of LRRK2, Beilina suggested.
A related presentation detailed a LRRK2 interaction that seems to clear the kinase from the cytoplasm. Frederick Nucifora in collaboration with Christopher Ross at Johns Hopkins University School of Medicine, Baltimore, Maryland, found with a yeast-two-hybrid screen and co-immunoprecipitation that both wild-type and mutant G2019S LRRK2 bound to an E3 ubiquitin ligase called WSB1. WSB1 and LRRK2 co-localized in lysosomes of cultured neuroblastoma cells. Evidence suggested that WSB1 tagged LRRK2 with ubiquitin, marking it for lysosomal destruction. Since boosting WSB1 prevented LRRK2 toxicity in cultured primary neurons, Nucifora proposed that activating the ligase to boost LRRK2 degradation may be a therapeutic strategy for PD.
In addition, Lewy bodies and Lewy neurites of postmortem human brains from sporadic PD patients contained WSB1, leading the team to hypothesize that the ligase encourages the formation of Lewy bodies to protect neurons in humans.
Still other evidence links LRRK2 function to nuclear organization, according to an October 17 Nature paper from Juan Carlos Izpisua Belmonte, Salk Institute for Biological Studies, La Jolla, California, and colleagues. Since aging correlates with nuclear envelope abnormalities, joint first authors Guang-Hui Liu, Jing Qu, and Keiichiro Suzuki wondered if diseases associated with aging also came with wrinkles in the nuclear architecture. They zeroed in on Parkinson’s and the G2019S mutation of LRRK2.
The researchers aged cultured human neural stem cells and found that those that express the mutant form of LRRK2 developed enlarged and convoluted (rather than spherical) nuclei with unusual folds in their nuclear membranes. The membranes lacked the filament proteins lamin B1 and lamin B2, which regulate nuclear structure. It turns out that these proteins were hyperphosphorylated relative to those in wild-type cells, suggesting that too much LRRK2 activation boosts phosphorylation of lamin B proteins.
Epigenetic changes to DNA accompanied the misshapen nuclear membranes, particularly affecting genes involved in neurogenesis and neural function. Furthermore, aged LRRK2 mutant cells divided and differentiated less well. These problems disappeared when the researchers introduced wild-type LRRK2 into the neural stem cells with a lentivirus. The results seemed to relate to disease pathology, since similarly deformed nuclei turned up in hippocampal cells from postmortem PD brains either with or without the G2019S mutation. Taken together, the results suggest involvement of the nucleus in PD pathology.
These proposed doings of LRRK2 add to previous evidence implicating it in synaptic vesicle endocytosis (see ARF related news story) and perturbation of the Golgi network (see ARF related news story). Unfortunately, no one has yet been able to finger the true pathogenic role of LRRK2. Several groups recently put together a consortium to study some 2,000 people with LRRK2 mutations worldwide (see ARF related news story).
"It could be that LRRK2 has myriad functions and activities, and only one or a few of them are pathological,” said Moore. "It’s going to be difficult to work out which one is most relevant to the disease."—Gwyneth Dickey Zakaib
No Available Comments
“Earlier” seems to be the new mantra in Alzheimer’s research. With biomarker data indicating that amyloid pathology begins up to 20 years before symptoms appear, many researchers now believe that treatment should start in the preclinical phase. This shift in thinking demands new ways to recognize the disease in its preliminary stages. In an Alzheimer’s disease press conference at the Society for Neuroscience 2012 annual meeting, held 13-17 October in New Orleans, Louisiana, researchers presented three novel approaches to early detection. These included using positron emission tomography (PET) to detect subtle functional changes a decade before diagnosis, imaging oligomeric forms of Aβ in living brains, and identifying an epigenetic signature of AD that distinguishes it from related disorders. Although promising, all of these strategies are under development and not yet viable as diagnostics. Two additional talks at the press conference focused on treatment, with researchers discussing insights gleaned from animal models (see Part 2).
With brain pathology accumulating up to two decades before diagnosis, Lori Beason-Held at the National Institute on Aging, Baltimore, Maryland, wondered whether changes in brain function would also be detectable at preclinical stages. To answer this, Beason-Held and colleagues looked at resting-state PET imaging of oxygen-15-labeled water to measure cerebral blood flow. About 120 people in the Baltimore Longitudinal Study of Aging participated. The study began while they were cognitively healthy, and continued with annual scans and cognitive tests. About one in six participants developed cognitive impairment, which surfaced an average of 11 years after the study began.
Over the first seven years of the study, the researchers saw significantly greater changes in blood flow patterns in people who developed memory problems compared to those who maintained good cognitive function. Prior to cognitive impairment, blood flow dropped in parietal and occipital cortices and the thalamus, but surged in several anterior brain regions such as the orbitofrontal and medial frontal cortex and the anterior cingulate. These frontal regions affect memory, attention, and executive function, all faculties that falter in AD, Beason-Held noted. Amyloid and tau pathology emerge early in affected brain regions as well, suggesting that changes in blood flow may reflect underlying pathology. Importantly, these changes take place several years before cognitive decline begins. Although the finding holds potential for flagging people at risk for decline, at the moment the changes only reach significance at the population level, Beason-Held said. In other words, researchers do not yet know what amount of blood flow change would signify increased risk for an individual. Ultimately, a combination of biomarkers may provide the best measure of those at risk for the disease, Beason-Held told the audience.
One difficulty in diagnosing AD is distinguishing it from other neurodegenerative diseases with overlapping symptoms, such as Parkinson’s disease and dementia with Lewy bodies. Paula Desplats at the University of California, San Diego, wondered if unique epigenetic alterations to DNA might characterize these diseases (see ARF related news story). To examine this, Desplats and colleagues compared frontal cortex autopsy samples from people with AD, PD, and DLB, as well as from healthy, age-matched controls. Each group contained about seven samples. The researchers focused on 84 genes that affect DNA structure, such as histone deacetylases (HDACs) and histone lysine methyltransferases. Several other studies have fingered HDACs and other epigenetic modifiers as therapeutic targets in AD (see, e.g., ARF related news story; ARF news story; and ARF news story).
The researchers found that 13 to 20 genes in each disease significantly changed expression compared to controls. While several genes showed similar changes in two or three of the disorders, about six unique genes also characterized each disease. The results provide a molecular signature of the three disorders, Desplats said. To make this into a useful biomarker, however, researchers will need to detect these changes in living people. If the epigenetic modifications occur systemically and not just in the brain, they might be detectable with a simple blood test, Desplats suggested. If so, clinicians could potentially use this method to help sharpen diagnosis and start patients on the correct treatment regimen earlier. The findings may also point to new therapeutic targets for each disease, Desplats noted.
Current imaging and fluid biomarkers provide glimpses into the early stages of AD, but these methods have some limitations, said William Klein at Northwestern University, Evanston, Illinois. For example, while scientists can visualize amyloid plaques in the brain using PET radioligands that bind to aggregated forms of Aβ (see, e.g., ARF related news story and ARF news story), plaque load correlates poorly with clinical symptoms, whereas synapse loss associates closely with cognitive decline (see, e.g., ARF related news story and ARF news story). Many researchers now believe that soluble, oligomeric forms of Aβ are most toxic to synapses and perhaps initiate the disease. Researchers need a way to visualize this synaptotoxic species, Klein said. He noted that the concentration of Aβ oligomers in AD brain shoots up to about seven times that of control brain before plaques form. This suggests that the presence of oligomers could make a good early diagnostic, Klein said.
To develop a probe, Klein and colleagues conjugated magnetic nanoparticles to antibodies specific for oligomeric forms of synthetic Aβ known as Aβ-derived diffusible ligands (ADDLs) (see ARF related news story and ARF news story). The resulting probe produces a strong magnetic resonance imaging (MRI) signal. In solutions and cell cultures, the compound binds to these Aβ oligomers with high affinity and specificity, Klein reported. The researchers incubated the probe with slices from human frontal cortex, and showed the MRI signal could clearly distinguish between AD and aged control brains.
Large molecules such as antibodies poorly penetrate the blood-brain barrier, presenting a problem for in-vivo use. To enhance uptake, the researchers delivered the antibody to transgenic AD mice via intranasal injections. In these experiments, they conjugated the antibody to a wheat germ protein rather than the magnetic nanoprobe. Treated mice were spared the cognitive declines seen in untreated animals, suggesting the antibody enters the brain and mops up Aβ oligomers, Klein said. The researchers are now working on a way to deliver probe-conjugated antibody to people by using a nasal spray. Preliminary experiments suggest that the probe enters the brain within six hours, allowing it to be used as a diagnostic. People at the press conference wondered how this diagnostic tool turns out to prevent cognitive decline in mouse models. Klein noted that the antibody is an example of “theranostics,” where the same agent could be used for both diagnosis and therapy. In addition, the probe could also evaluate the efficacy of early-stage drugs designed to lower Aβ levels, Klein suggested. Other groups are developing similar theranostics that recognize oligomeric forms of Aβ (see ARF related news story).—Madolyn Bowman Rogers.
This is Part 1 of a two-part series. See also Part 2.
Animal models of Alzheimer’s disease have provided key insights into the disorder, but researchers agree they have limitations for studying human disease. At the Society for Neuroscience 2012 annual meeting, held 13-17 October in New Orleans, Louisiana, several presentations raised questions about current animal models and their relationships to disease mechanisms. For example, researchers reported that in mice, overexpression of amyloid precursor protein (APP) during development—something that does not occur in human disease—blunts the ability of the adult brain to respond to anti-amyloid therapy. This hints that current transgenic animal screens could miss useful drugs. Other presentations fueled doubts about the toxicity of Aβ42, looked at the effects of mouse background strain on amyloid pathology, and described an unusual strategy for targeting tau pathways. Several talks noted that amyloid plaques by themselves are insufficient to weaken cognition in mice, which seems true in people as well (see, e.g., ARF related news story; ARF news story). Two of the studies were chosen for discussion at an SfN press conference on AD, while others were part of nanosymposia on animal models and Aβ (see Part 1 of this series for other press conference coverage).
In most amyloid-based models of AD, mutant gene expression occurs throughout the life of the animal. This may not reliably reflect human AD, in which Aβ deposits accumulate only late in life, said Alena Savonenko at Johns Hopkins University, Baltimore, Maryland, in the press conference. She wondered what would happen if APP overexpression, a mainstay of most mouse mimics of AD, turned on only in adulthood. To explore this, she used the TetO-APPSwe/Ind mouse created in 2005 by Joanna Jankowsky, then at David Borchelt’s lab at Johns Hopkins (see Jankowsky et al., 2005). In this animal, the human APP transgene is under the control of the TetO promoter and can be turned off by feeding the mice doxycycline (see description of Tet-Off System). The scientists first used the model to test what happened when they shut down APP production late in life, after plaques had formed (see ARF related news story). Savonenko, who was a coauthor on the earlier paper, tried the opposite approach. She turned off the transgene during development to more closely mimic the human situation.
Savonenko and colleagues fed half the TetO-APP mice doxycycline for their first month of life to suppress the APP gene (these mice are called late expressors), while the other half produced APP throughout development (early expressors). After the first month, all mice were allowed to express the transgene. At 13.5 months old, both groups had similar plaque loads and levels of soluble Aβ in their brains, and both showed impaired spatial memory in the Y-maze, Savonenko reported. The researchers then switched off APP for one week in all mice to simulate the effects of anti-amyloid therapy given late in life. Plaque burden did not change, but in the late expressors, spatial memory bounced back to wild-type levels, while the early expressors showed no improvement. This implies that the presence of APP or one of its metabolites during development weakens the ability of the adult brain to respond to anti-amyloid treatment, Savonenko told the audience. Since most current APP mouse models overexpress the transgene throughout life, they might miss the effects of experimental drugs, she said.
The researchers saw other differences. Early expressors became more hyperactive than late expressors, although both groups returned to normal activity levels after APP was shut off. The brains of the early group may become sensitized to the effects of APP, said Jankowsky, who is now at Baylor College of Medicine, Houston, Texas, in a nanosymposium talk. She noted that when the transgene stays silent for the first six weeks of life, adult APP mice have normal activity levels (see Rodgers et al., 2012). This points to a developmental effect of APP on motor circuits that control activity, she suggested. Early expressors also show abnormalities on electroencephalograms, displaying sharp discharges that get worse with age. Late expressors, by contrast, look normal by EEG. Other researchers have found abnormal EEG activity linked to seizures in some strains of AD mice (see ARF related news story).
In a separate talk, Borchelt, now at the University of Florida, Gainesville, discussed whether APP or one of its breakdown products causes the observed cognitive and behavioral impairments in TetO-APP mice. In one experiment, he gave 13-month-old mice doxycycline to turn off APP expression, and their cognition rapidly improved compared to control animals. Borchelt ruled out relief from Aβ monomers as the cause of the improvement, noting that their levels stayed the same whether mice were on or off doxycycline, even after four weeks of treatment. He believes that soluble Aβ oligomers are not culpable, either, since he could not detect them by Western blotting of extracts from TetO-APP mice either on or off doxycycline, even using antibodies 6E10 and 4G8, which are sensitive to nanogram quantities. Oligomers do remain in the insoluble membrane fraction, but show no change on or off doxycycline, he added. By contrast, APP, its C-terminal fragments (CTFs), and sAPPα and β levels all drop in animals on doxycycline, correlating with memory improvements. The results suggest that sAPP, APP, or CTFs could be more than just bystanders in cognitive decline, and might interact with Aβ to weaken cognition, Borchelt said. Other researchers have found evidence for APP/Aβ interactions (see ARF related news story) and β-CTF toxicity (see ARF related news story; ARF news story; and ARF news story), or have pointed to a pyroglutamate-modified form of Aβ as the bad seed (see ARF related news story).
In the same nanosymposium, Chris Janus at the University of Florida, Gainesville, also raised the question of the toxic Aβ entity. He used BRI-Aβ42 mouse models created by Todd Golde and colleagues at the Mayo Clinic in Jacksonville, Florida (see ARF related news story on McGowan et al., 2005). These animals express Aβ42 fused to the BRI protein. The enzyme furin cleaves off Aβ and releases it into the extracellular matrix without the need for APP overexpression or processing. The mice deposit Aβ and develop florid plaques in forebrain by 17 months of age. Nonetheless, they show no behavioral impairments, Janus reported. He tested them on a wide variety of assays, including fear conditioning, open field anxiety tests, rotarod tests of motor coordination, water maze memory tests, and conditioned taste aversion, but found no abnormalities. Chronic exposure to aggregated extracellular Aβ by itself is not sufficient to cause cognitive decline, Janus concluded. The findings echo those in the TetO-APP mice, and even in humans, where plaques do not seem to directly affect cognition. An audience member suggested that Aβ localization might make a difference, pointing out that in traditional mouse models, Aβ gets released at synaptic clefts in response to activity, putting it in the right place to harm transmission. Further muddying the waters, a rat model that features viral overexpression of BRI-Aβ42 and/or BRI-Aβ40 in the hippocampus does show cognitive impairments (see Lawlor et al., 2007).
Other researchers suggested that strain differences confound AD mouse models. Takashi Morihara at Osaka University, Japan, leveraged such differences to find genes that interact with APP to modify pathology in the hopes that this might cast light on causes of sporadic AD. He crossed Tg2576 mice, which express APP with the Swedish mutation, into three backgrounds: C57BL/6, SJL, and DBA/2. While APP expression remained the same on all backgrounds, DBA mice had lower levels of cortical Aβ42 than did the other two strains.
To identify genes responsible, Morihara and colleagues used a transcriptomics approach to compare RNA expression from the three strains. Among the 54 genes that varied among the mice, four showed changes in expression that correlated with Aβ levels. Two of those candidates were splice variants of kinesin light chain 1 (KLC1). This protein associates with microtubules and plays a role in organelle transport and trafficking from the Golgi apparatus. Morihara found that DBA mice produce less KLC1 variant E. This splice form modifies Aβ accumulation, Morihara concluded. He speculated that variant E might affect APP localization and, thus, processing. Other work has linked Aβ production to APP localization within endosomes (see, e.g., ARF related news story and ARF news story). Mouse KLC1 splice patterns are conserved in people, Morihara said. He found increased levels of KLC1 variant E mRNA in AD brains, suggesting that in humans, too, it may contribute to Aβ burden.
In a change of pace from these Aβ-centered talks, Fred Van Leuven at KULeuven, Belgium, described a tau-based treatment strategy. At the press conference, he pointed out that although phosphorylation gets the lion’s share of the attention, tau can undergo several types of modifications that might affect its behavior. Van Leuven focused on O-GlcNAcylation, the addition of a sugar molecule to a protein. Other groups have reported that O-GlcNAcylation of tau and several other proteins drops in the Alzheimer’s brain (see, e.g., Liu et al., 2009; Dias and Hart, 2007; Robertson et al., 2004), and that pumping up O-GlcNAcylation can slow neurodegeneration in tau model mice (see Yuzwa et al., 2012, and Yu et al., 2012).
Van Leuven and colleagues tested this idea in aged P301L tau mice and in young biAT mice, which express mutant human APP as well as tau, and typically die at four months old (see Terwel et al., 2008). To accelerate O-GlcNAcylation, the researchers fed the animals an inhibitor of O-GlcNAcase, the enzyme that removes sugars from proteins. In agreement with their hypothesis, treated animals had significantly better motor skills, higher body weight, and longer lifespan compared to controls. The surprise came when the researchers isolated tau from the mouse brains, and found no sugars on it in either treated or untreated mice. This means tau is not O-GlcNAcylated in mouse brain, Van Leuven concluded. The finding conflicts with reports of O-GlcNAcylated tau in P301L-JNPL3 mice, which express a slightly different form of mutant tau (see Yuzwa et al., 2012). The data suggest the drug acted on some other protein, Van Leuven said. About 250 proteins get O-GlcNAcylated in mouse brain, and he speculated that the culprit probably lies downstream of tau in the pathological cascade. He is now looking for the target, and believes it will provide insight into neurodegenerative pathways downstream of tau.—Madolyn Bowman Rogers.
This is Part 2 of a two-part series. See also Part 1.
No Available Comments
Neurons are a minority of cells in the brain, with glia, including microglia and astrocytes, dominating. It may seem due time, then, that that majority is drawing the attention of Alzheimer’s and other neurodegenerative disease researchers. The topic stood out as a theme at the Society for Neuroscience 42nd annual meeting, held 13-17 October in New Orleans. Scientists are slowly beginning to understand the interplay between glial-neuronal crosstalk and AD pathology, though much more work needs to be done.
Glia and Inflammation
The interplay between glia and Aβ is one aspect of AD pathology that continues to mystify. Although researchers know that glia mop up Aβ deposits, and that the peptide can render the cells proinflammatory and potentially toxic (see ARF related news story), the signals involved are poorly understood. Toll-like immune receptors (TLRs) on microglia seem to have a hand in this (see ARF related news story on Reed-Geaghan et al., 2009), but knocking out individual receptors has not painted a clear picture. Gary Landreth at Case Western Reserve University, Cleveland, Ohio, explained that TLRs are part of a very large molecular complex that fails to form properly when the receptors are absent, confounding interpretation of the knockout phenotypes.
Landreth’s group took a different approach. Instead of targeting TLRs, they went for interleukin receptor-associated kinase 4 (IRAK4), a member of a family of kinases that mediate signaling through the TLR complexes. As presented on a poster, first author Brent Cameron and colleagues replaced the normal mouse gene for IRAK4 with one that codes for an inactive form. “While knocking out TLRs and co-receptors ends up disrupting the complex, using this knock-in strategy we allow the complexes to form naturally, but they cannot signal,” Landreth told Alzforum.
Cameron knocked the inactive IRAK4 into APP/PS1-21 mice obtained from Mathias Jucker’s lab at the University of Tubingen, Germany. These mice aggressively deposit Aβ beginning at about six weeks of age. The researchers found that Aβ pathology in IRAK4-negative APPPS1 mice and their kinase-competent littermates seemed the same at four months of age. By eight months, however, the IRAK4 mutants accumulated 40 and 30 percent less soluble and insoluble Aβ42, respectively, in the brain. They also had fewer and smaller amyloid plaques (see Cameron et al., 2012).
How does loss of the kinase signal translate into reduced Aβ? The researchers looked to inflammatory responses that might exacerbate Aβ production. But instead of finding a reduction in proinflammatory markers, they found that microglia from IRAK4-negative eight-month-old transgenics expressed higher amounts of both pro- and anti-inflammatory markers compared to APP/PS1 controls. “We interpret this to mean that pro-inflammatory microglia may be associated with plaques, but a few millimeters away there are glia that are not involved,” said Landreth. That suggests that loss of IRAK4 renders microglia generally more quiescent, while still allowing them to respond to perturbances in their environment, such as plaques.
Support for this idea came from analysis of interferon response factors (IRFs). These transcription factors are master regulators of toll-like receptor responses. Recently, scientists discovered that IRF4 and IRF5 have opposing actions on microglia. IRF4 promotes an anti-inflammatory (M2) state (see Ahyi et al., 2009), while IRF5 induces pro-inflammatory (M1) responses and suppresses M2 responses (Krausgruber et al., 2011). Cameron and colleagues found that, beginning at four months, the IRAK4-deficient animals began to switch to an M2 state. They made 75 percent less IRF5 at four months, and by eight months, 40 percent more IRF4 and 55 percent less IRF5. While the data could be interpreted to mean that loss of toll-like receptor signaling favors the M2 state, Landreth admitted that the story is complex. The challenge in determining the role of glia in pathology lies in understanding their heterogeneity, he suggested. “Microglia only care about what is happening in their vicinity,” he said. “Until we get the right tools, such as better histochemistry or single-cell analysis, we will not be able to understand that complexity.”
Kiran Bhaskar, now at the University of New Mexico, Albuquerque, addressed a different type of glial signaling. When he worked with Bruce Lamb at the Cleveland Clinic in Ohio, Bhaskar reported that fractalkine, a peptide released by neurons, seems to quench glial inflammatory feedback on those same neurons. Fractalkine activates the glial CX3CR1 receptor. When the researchers crossed CX3CR1 knockout mice with animals expressing human mutant tau, hyperphosphorylation and aggregation of tau ran rampant in response to an inflammatory stimulus. Neurodegeneration rose as well (see ARF related news story). The findings suggested that fractalkine signaling keeps tau pathology in check. On the flipside, Bhaskar predicted that tau pathology would exacerbate inflammatory glial signaling.
In New Orleans, he showed what happens when he took tau out of the picture. Knocking out tau protected primary neurons against activated microglia. Lipopolysaccharide (LPS), a potent inducer of glial inflammation, triggered cell death in mutant human tau-positive neurons co-cultured with CX3CR1-negative microglia, but tau-negative neurons were mostly unaffected. Neurons lacking tau showed little increase in the apoptosis markers caspase 3 and annexin V in comparison to control neurons. Similarly, in two-month-old tau-negative mice, only half as many cells expressed caspase 3 in the dentate gyrus as in tau-positive animals. Furthermore, LPS barely activated microglia in tau knockout mice, as judged by staining with the glial marker Iba1. The cellular protection afforded by knocking out tau seemed to translate into behavioral gains. LPS-treated CX3CR1 knockout mice spent more time in the open field than wild-type animals, indicating a lack of inhibition, but on removal of tau the animals mostly avoided the open, just like wild-type mice. All told, the work suggests that tau supports inflammatory crosstalk between neurons and glia.
Bhaskar is unsure why knocking out neuronal tau prevented microglial activation, especially since there should be no fractalkine signaling between the neurons and the CX3CR1-negative glia. “At this point, I think communication between neurons and microglia is independent of CX3CL1-CX3CR1 signaling, but may be dependent upon other possible signaling pathways,” he told Alzforum (e.g., see Ransohoff and Cardona, 2010). Bhaskar also said that other cell markers of glial activation depended on neuronal tau. He plans to explore other neuron-glial signaling pathways that might depend on tau.
Astrocytes and Neurotransmission
Astrocytes are the other major glia in the brain. They, too, respond to Aβ. At SfN, researchers led by Rheinallt Parri at Aston University, Birmingham, U.K., and Kelly Dineley at the University of Texas, Galveston, presented data showing that Aβ acts on astrocytic acetylcholine receptors to boost release of glutamate, which then signals to neurons. First, Parri showed that acute hippocampal slices from young, nine- to 14-day postnatal Tg2576 mice generated more spontaneous astrocyte calcium spikes than slices from wild-type animals. Adding Aβ1-42 (300 pM) to wild-type slices increased astrocytic activity, and that boost was blocked by an α7-nicotinic acetylcholine receptor (α7-nAChR) antagonist. On the other hand, adding Aβ1-42 did not further increase activity in transgenic slices, indicating that the receptors were already saturated. Parri believes that astrocyte activity releases glutamate, which can then activate adjacent neurons. Patch-clamp recordings from hippocampal neurons revealed slow inward currents (SICs) consistent with glial glutamate transmission to neuronal NMDA receptors. Inward currents in Tg2576 slices persisted for longer than in wild-type tissue and could be induced by an α7-nAChR agonist. Parri claims that increased astrocytic activity in the Tg2576 animals leads to enhanced gliotransmission. Since others have proposed that Aβ production occurs with synaptic activity, these results suggest a potential role for endogenous Aβ to modulate glutamate neurotransmission via interaction with astrocytic α7-nAChRs, suggested Dineley. “The take-home message from this work is that astrocyte gliotransmission becomes dysfunctional early, before any effect on cognitive function,” she said.
Researchers from South Korea looked to a different transmitter in astrocytes—γ-aminobutyric acid. On her poster, Seonmi Jo, from C. Justin Lee’s lab at the Korea Institute for Science and Technology, Seoul, reported finding high levels of GABA in reactive astrocytes in APPswe/PS1 transgenic animals. Jo used a GABA-specific antibody to detect a fivefold increase above astrocytic levels in wild-type mice. Interestingly, astrocyte glutamic acid decarboxylase and GABA transporters appeared normal in the transgenic mice, apparently ruling out uptake or synthesis from glutamic acid as explanations for GABA increases. Instead, the researchers found elevated putrescine and monoamine oxidase, which can metabolize the polyamine to GABA. Closing the loop, they found that the astrocytes from the transgenic animals lacked the transaminase that normally metabolizes GABA.
The data suggest a buildup of GABA in astrocytes in the APP/PS1 mice. How might this unconventional finding fit with AD pathology? Using in-situ microdialysis, Jo and colleagues detected 60 percent more GABA in the dentate gyrus of the transgenics, as well as tonic inhibition by GABA. A GABA receptor antagonist boosted neural transmission in the transgenic animals but not in wild-type mice. The researchers were unable to show that the elevated GABA impairs learning and memory in these transgenic mice. Even so, they suggested that astrocytic GABA might be worth exploring as a therapeutic target in AD.—Tom Fagan.
No Available Comments
Franklin’s genius notwithstanding, death and taxes are not the only things that are certain. It’s pretty clear you can’t evade your genetics, either. But what about epigenetics? While tweaking the tax code might improve your fortune, changes that alter the outcome of your genetic code could improve your health. At the 42nd annual meeting of the Society for Neuroscience, held 13-17 October in New Orleans, Louisiana, researchers pored over epigenetic marks predisposing to AD and cognitive decline, and discussed how those might be targeted therapeutically. While the work seems promising, scientists cautioned that these are still early days, and many questions remain difficult to answer. “Epigenetics is a newly developing field, and much remains to be worked out,” said Paul Coleman, Banner Sun Health Research Institute, Sun City, Arizona. “Nevertheless, I believe it will turn out to be at least as important as genetics itself,” he predicted. With Suzana Petanceska from the National Institute on Aging, Bethesda, Maryland, Coleman co-chaired a mini-symposium on the role of epigenetics in the development and maintenance of human cognition.
Unbiased Approaches
Many research labs, including Coleman’s, investigate links between disease and DNA methylation, which typically occurs on cytosine-guanine dinucleotide (CpG) sequences. Methylation silences genes and can profoundly alter biology (see ARF related news story). However, faced with upwards of 20 million CpG sites in the human genome, how are scientists to finger those that are most important to the brain? At the SfN meeting, David Bennett, Rush University Medical Center, Chicago, Illinois, outlined a quantitative trait strategy to identify some that influence AD pathology and/or cognitive decline.
Bennett and colleagues used samples from the Religious Orders Study and the Rush Memory and Aging Project. These two longitudinal observational studies correlate dementia incidence with epidemiological and pathological data. They probed DNA samples taken from the postmortem dorsolateral prefrontal cortex with an Illumina beadset that detects methyl groups at ~485,000 sites in the human genome. Bennett reported that methylation on many of those sites is coordinated, i.e., when a given site is methylated, so are others nearby. The number of such methylation blocks, or mBlocks, in the whole genome could top 300,000 he said.
Turning to AD, Bennett reported significant correlations between methylation at 168 sites in the genome and the presence of neuritic plaques seen on autopsy of 759 subjects. The methylation site that most tightly associated with plaques does not lie near any known genes, but the next two strongest sites are close to each other and to the AGPAT6, ANK1, and NKX6-3 genes. None of those have been previously linked to AD. Two of the 168 methylation sites turned up in AD susceptibility loci indentified in recent genomewide association studies: BIN1 and ABCA7, which rank second and fourth in the AlzGene database, respectively. Looking at pathologic cases and controls, Bennett predicted that about 45 percent of the variance in plaque burden can be explained by methylation among the 168 sites.
Are the genes near any of these methylation sites up- or downregulated in AD? Using RNA microarrays to correlate gene expression with plaque burden in the Religious Orders and the Rush Memory and Aging samples, Bennett found in eight of the 168 loci there were eight genes with expression patterns that correlated with neuritic plaque burden. Assessing those eight genes against an independent set of samples from the Mayo Clinic, where four of the genes—AP3M2, ACACB, DDB1, and HSPB2—were differentially expressed in the hippocampus of AD samples compared to controls. HSPB2 expression was linked to astrocytes in the vicinity of neuritic plaques before, but the other genes had not been previously associated with AD. None appears to be linked to AD; however, a separate analysis that looked for correlation with cognitive decline revealed a methylation site near the α-2 macroglobulin gene, which has been genetically linked to AD in some studies (see AlzGene entry).
Dana Dolinoy and colleagues at the University of Michigan, Ann Arbor, adopted a similar approach but with smaller sample sets. These researchers probed postmortem frontal cortex samples from 12 late-onset AD patients and 12 cognitively normal age- and sex-matched controls. They used a smaller Illumina array that surveys ~28,000 CpG sites spanning almost 15,000 genes. At the SfN meeting, Dolinoy reported that, overall, she found no dramatic differences in methylation status between the AD and control samples. In both, there was a cluster of sites that was between 75 and 100 percent methylated, and a second cluster with less than 10 percent methylation. Dolinoy said that 948 CpG sites might be potentially associated with AD, while over 2,400 associated with age. She showed a “heat map” for the top 25 that indicated both hypo- and hypermethylated sites in the AD samples. Honors for being most strongly associated with AD went to a CpG in the promoter of the transmembrane protein 59 gene (TMEM59), which is reported to modulate expression and processing of APP (see Ullrich et al., 2010). The researchers found that TMEM59 methylation associated with AD in an additional three matched sample pairs. RNA and protein levels also correlated to methylation status (see Bakulski et al., 2012).
Focused Attempts
Researchers agreed that these unbiased approaches for scanning the genome have their shortcomings. For example, the arrays only scan a fraction of the number of potential methylation sites. They need a large amount of starting material, which is a problem because it means scientists have to use homogenates rather than defined cell types or single cells, said Coleman.
Philip Landfield’s group at the University of Kentucky, Lexington, has used laser capture microscopy to isolate and study single cells for transcriptional changes in AD tissue. Up- or downregulated gene expression could reflect epigenetic changes that predispose to disease, he said. Previously, Landfield’s group used microarray analysis to look for correlations between hippocampal gene expression and cognition or AD pathology. The scientists reported that several thousand genes may be regulated up or down as Mini-Mental State Exam (MMSE) scores declined, 89 of which correlated with MMSE score and neurofibrillary tangle burden (see ARF related news story). While that analysis relied on whole tissue sampling, Landfield reported at the SfN meeting that his group has revisited that analysis using laser capture to isolate grey matter from the CA1 region of the hippocampus (see Blalock et al., 2011). This time the scientists did not detect correlations with glial and growth factors that had been positive in the previous study, suggesting that those AD-related changes are specific to white matter. “The functional relevance of these findings remains a major question,” said Landfield. Genes involved in chromatin assembly and organization, including histone acetyltransferases and histone deacetylases, were upregulated in samples from people with incipient AD. The authors found that similar changes to the same epigenetic regulators occurred with normal aging. In a variation on the classic "who will guard the guards," Landfield noted that "the altered expression of epigenetic regulators in the hippocampus with aging and/or AD indicates that they are, in turn, modified by other unknown factors."
Other research groups are also taking more direct approaches. Farah Lubin and colleagues at the University of Alabama at Birmingham have looked at histone methylation to see if it plays a role in memory consolidation. Transcription drives long-term memory, and histone modifications can activate or repress genes (see ARF related news story on Gupta et al., 2010). Lubin decided to examine the involvement of the histone lysine methyltransferase G9a. This enzyme methylates lysine 9 on histone H3, which in turn recruits other proteins that repress transcription, including histone deacetylases (HDACs). There are indications that G9a may be important in some forms of plasticity associated with cocaine addiction (Maze et al., 2010), but whether it more broadly influences learning and memory was not clear.
Hints came from studying changes elicited by contextual fear conditioning, a type of learning that involves crosstalk between the hippocampus and the entorhinal cortex. At SfN, Lubin showed that a foot shock drove methylation on lysine 9 of H3 (H3K9) in this brain area. To test if the methylation was important for learning, she blocked the methyltransferase by injecting an inhibitor (BIX01294) into the brain an hour before fear conditioning. Animals given CA1 injections of BIX01294 froze half as often as did untreated mice when reintroduced into the conditioning environment a day later. This suggests that the methylase supports this type of learning, Lubin said. In contrast, injecting the methylase inhibitor into the entorhinal cortex boosted the animals’ memory of the foot shock. These opposite effects revealed a complex relationship between this epigenetic modification and memory, said Lubin. In keeping with this, the methylase inhibitor suppressed long-term potentiation (LTP) in the Schaeffer collateral pathway of the hippocampus. Interestingly, blocking G9a in the ERC raised methylation in the hippocampal CA1.
How might methylation work in this context? Lubin showed that BIX01294 infused into the ERC elevated histone methylation at the promoter for the catechol-O-methyltransferase (COMT) gene in the CA1. Despite its name, COMT is not involved in DNA or histone modification, but instead metabolizes catecholamines, including dopamine, which support long-term memory. This finding shows that epigenetic changes to histones, which could influence many different genes, can directly modulate genes involved in memory.
Stephen Haggarty, Massachusetts General Hospital, Boston, also spoke to the role of epigenetics in plasticity. Working with Li-Huei Tsai, at MIT, Haggarty and colleagues work to tease out the roles of HDACs in learning and memory. The brain contains many different HDACs. The researchers found earlier that HDAC2 suppresses memory in mice (see ARF related news story), and that this deacetylase is activated in mouse models of neurodegeneration (see ARF related news story). The researchers are looking for compounds to block HDAC2, though its similarity to HDAC1, which is neuroprotective, makes this difficult, Haggarty pointed out (see ARF related news story on Kim et al., 2008). Using combinatorial chemistry, Haggarty’s collaborators built a family of macrocyclic compounds (see Marcaurelle et al., 2010) and tested them for HDAC inhibition in an unbiased screen. Of 14 hits, none was selective against HDAC2. Haggarty reported some success in screens for HDAC1 activators. Prototypes also activated HDAC2, but only at higher concentrations, said Haggarty, raising hopes that specific HDAC1 activators can be developed.
Looking to a different therapeutic approach, Claes Wahlestedt from the University of Miami, Florida, talked about using antagoNATs, or antagonists to natural antisense transcripts. Wahlestedt pointed out that for many human genes, both sense and antisense transcription goes on at the same time; hence, the antisense transcript can potentially repress transcription of the sense strands. Scientists are still working out how this repression occurs, but it seems to involve epigenetic regulation, namely, modification to chromatin histones. Blocking antisense transcripts could, therefore, relieve this type of transcriptional repression, said Wahlestedt.
As an example, the Miami group focused on regulation of brain-derived neurotrophic factor. BDNF protects neurons from degeneration, and its loss with age may put people at risk for AD (see ARF related news story). The brain makes antisense BDNF strands, and Wahlestedt reported that antagoNATs to those transcripts boost expression of the sense transcript. Working with collaborators, he generated several BDNF antagoNATs using locked nucleic acids. These are chemical analogs that resist degradation by nucleases. When the scientists added these antagoNATs to N2a neuroblastoma cells, they saw a robust increase in sense BDNF transcripts (see Modarresi et al., 2012). The strategy worked in vivo, too. Delivered by mini-osmotic pumps into the brain ventricles over four weeks, the antagoNATs drove up BDNF mRNA and protein across the forebrain. This increased neuronal survival and proliferation.
Wahlestedt said this antagoNAT strategy could be useful for neurodegenerative diseases, claiming the gains may go beyond BDNF. Of 250 different genes Wahlestedt looked at, 132 have natural antisense transcripts.
Overall, the symposium highlighted the angles researchers are taking to examine the role of epigenetics in normal brain function and in disease, from genomewide to candidate approaches. How do the pieces fit into a comprehensive model of learning, memory, and neurodegeneration? “That is a key challenge in the field,” said Coleman.—Tom Fagan.
No Available Comments
A growing number of studies stress the harmful effects of tau in Alzheimer’s disease, but exactly how the protein exerts toxicity, and how it interacts with Aβ, remain open to question. At the Society for Neuroscience 42nd annual meeting, held 13-17 October in New Orleans, Louisiana, speakers offered new clues during a nanosymposium on tau and Aβ. One talk focused on extracellular tau and its role in propagating disease, while two other researchers reported mechanistic links between Aβ and tau via cell cycle proteins and proteases, respectively.
Irene Griswold-Prenner from the biotech company iPierian in South San Francisco, California, addressed the spread of tau pathology from cell to cell. Other researchers have suggested this occurs by tau secretion and templated protein misfolding (see ARF related news story; ARF news story; ARF related news story; and Lasagna-Reeves et al., 2012). To investigate the mechanisms, Griswold-Prenner and colleagues first made a cellular model of AD. They took skin cells from familial AD patients and people with Down’s syndrome and turned them into induced pluripotent stem cells (see ARF related news story; ARF news story; and ARF related news story). Then they differentiated the cells into cortical neurons.
As expected, induced neurons made from people with presenilin 1 (PS1) and PS2 mutations had higher Aβ42/Aβ40 ratios compared to control cells, although they did not show a difference in total Aβ, Griswold-Prenner said. More surprising was the finding that the induced FAD neurons secreted up to twice as much extracellular tau (e-tau) as did control neurons. Although unexpected, this finding is in keeping with the high cerebrospinal fluid (CSF) tau levels seen in AD patients, Griswold-Prenner told Alzforum.
What might the extracellular tau be doing? To investigate, the researchers added conditioned medium from these cultures to mouse cortical neurons. They saw rapid uptake. This seems to occur through active transport, as the process can be influenced by temperature, Griswold-Prenner noted. Treating cultures with cytochalasin D, an inhibitor of actin polymerization, also prevented e-tau internalization, again suggesting this is an active process perhaps mediated by endocytosis. Neurons that took up e-tau showed increased neuronal depolarization and hyperexcitability. Hyperactive neurons are another hallmark of AD, although it has usually been associated with Aβ (see ARF related news story; ARF news story).

A mouse cortical neuron (red) internalizes extracellular tau (green dots). Image courtesy of Bonnie Cooper, iPierian
What exactly is e-tau? Griswold-Prenner reported that it consists of fragments rather than full-length protein. In response to questions from the audience, she said she is not yet certain what parts of tau are in the extracellular forms. She noted that full-length tau had no effect on excitability in her stem cell-derived neurons.
Griswold-Prenner and colleagues also looked for e-tau in CSF from familial and sporadic AD patients and healthy controls using several commercial assays. As expected, they found elevated tau in AD patients, but standard assays cannot determine whether the protein is full length or fragmented, Griswold-Prenner said. To examine this, they isolated tau from pools of CSF using antibodies and looked at size on a Western blot. At SfN, Griswold-Prenner reported that the scientists consistently see tau fragments in CSF, but cannot detect any full-length tau. She is not sure yet if these fragments are the same as the ones released by the iPSC-derived neurons. Eckhard Mandelkow at the German Center for Neurodegenerative Diseases (DZNE) in Bonn, Germany, questioned the finding of fragmented CSF tau, noting that Kaj Blennow at the University of Gothenburg in Molndal, Sweden, reports full-length tau in CSF (see Portelius et al., 2008). However, the Swedish study used mass spectrometry, which relies on proteolytic digestion, and so could not directly confirm full-size tau, Griswold-Prenner told Alzforum.
Griswold-Prenner said iPierian has developed an antibody against e-tau that shows good drug properties and prevents e-tau toxicity in culture. The researchers are starting preclinical prevention studies with this agent in mouse models of frontotemporal dementia (FTD). In humans, several forms of FTD are pure tauopathies (see ARF related news story; ARF news story; ARF news story).
Another pressing question in the field is how tau relates to Aβ. Previous work in mice has shown that deleting tau prevents Aβ toxicity (see ARF related news story). Some studies reported that dendritic tau mediates Aβ excitotoxicity at NMDA receptors, suggesting a possible mechanism that connects the two proteins (see ARF related news story and ARF news story). Researchers led by George Bloom at the University of Virginia, Charlottesville, came to the Aβ/tau relationship from an entirely different direction. They were intrigued by work from Karl Herrup, then at Rutgers University, Piscataway, New Jersey, who showed that in AD, mature neurons attempt to re-enter the cell division cycle. These cells replicate their DNA, but then get stuck, unable to complete mitosis. After about a year, they die. Herrup and others recapitulated the effect by exposing primary cultured neurons to Aβ (see also ARF related news story). Herrup proposed that this phenomenon accounts for a large fraction of the neuronal loss in AD (ARF Webinar).
Bloom wondered what role tau might play in this process. As he described at SfN, his group first repeated Herrup’s results, treating primary neuronal cultures with either monomers or oligomers of synthetic Aβ. The latter induced DNA synthesis, as seen by the incorporation of the synthetic nucleoside bromodeoxyuridine (BrdU). Neurons from tau knockout mice, however, did not take up the nucleoside after Aβ treatment, indicating that tau is required for the cell cycle reentry effect. The researchers saw a similar phenomenon in vivo. In six-month-old hAPP J20 mice, nearly 60 percent of neurons express the cell cycle marker cyclin D1; however, when the J20s were crossed with tau knockout mice, cyclin D1 was undetectable in six-month-old offspring.
Investigating the mechanism, Bloom found that synthetic Aβ oligomers activate the kinases CaMKII, PKA, and Fyn, which each phosphorylate tau at specific sites (serine 416, 409, and tyrosine 18, respectively). Inhibiting one of these kinases, or mutating any of their target phosphorylation sites, prevented DNA replication after Aβ treatment. By contrast, inhibiting GSK-3β, a different tau kinase, did not stop this process. The results suggest that Aβ activates tau toxicity through CaMKII, PKA, and Fyn acting in parallel, with all three being essential to trigger cell cycle reentry, Bloom said. He noted that this mechanism occurs independently of any aggregation of Aβ or tau, fitting with a wealth of other data showing that the soluble forms of these proteins are the most deadly.
Other scientists are proposing different links between the two hallmark proteins of AD. For example, Samer Abdul-Hay at the Mayo Clinic, Jacksonville, Florida, reported at SfN that Aβ and tau are connected through the activity of a protease that degrades both. Abdul-Hay investigated amyloid-degrading proteases because late-onset AD is widely seen to result from the brain’s failure to clear Aβ. The aspartyl protease cathepsin D (CatD) has been genetically linked to AD, suggesting it might play a role in some cases (see ARF related news story), but the association is weak.
Abdul-Hay found that total insoluble (but not soluble) Aβ levels in the brains of CatD homozygous knockout mice rise until the animals die at about four weeks old. Amyloid accumulation topped that seen in knockouts of other amyloid-degrading enzymes, such as neprilysin or insulin-degrading enzyme, suggesting that CatD plays a more central role in degrading amyloid, Abdul-Hay said. Looking at the dynamics, Abdul-Hay found that CatD degrades Aβ40 most efficiently. In contrast, though Aβ42 binds the enzyme strongly, it also disengages slowly, making for slower proteolysis and for competitive inhibition of the protease. This means that the presence of high levels of Aβ42 can bog down CatD, allowing more amyloid to accumulate, he suggested. To further test CatD’s role, the researchers crossed CatD heterozygotes with Tg2576 mice, which carry mutant human APP. The offspring, who live to about six months, developed early, robust, intra- and extracellular amyloid pathology and huge amyloid plaques (50-80 microns in diameter) compared to their Tg2576 parents.
Abdul-Hay wondered if CatD downregulates tau as well. He used RTg4510 mice, which express human mutant tau under an inducible promoter, and introduced a viral construct to overexpress or downregulate the cathepsin. Overexpression chewed up tau, whereas inhibiting the protease slowed tau breakdown and increased its hyperphosphorylation. In toto, Abdul-Hay claimed that cathepsin D links Aβ and tau pathology indirectly, in that Aβ42 shuts down CatD activity, thus pumping up tau levels.—Madolyn Bowman Rogers.
No Available Comments
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.