Genetic Drivers for Cerebrovascular and Alzheimer’s Diseases Are Distinct
Quick Links
Cerebrovascular disease commonly occurs alongside Alzheimer’s. The two share some pathological features, such as leakiness of the blood-brain barrier. Do they share genetic drivers, too? No, according to scientists led by Alexi Nott at Imperial College London. As reported in the October 30 Neuron, the authors profiled the epigenome of human brain and cerebrovascular cells to find out which cell types expressed genetic variants linked to AD or to cerebral small vessel disease (SVD). Bottom line: Each disease had its own genetic signature.
- In human brain tissue, genes linked to white-matter hyperintensities are expressed mostly in astrocytes.
- Genes linked to enlarged perivascular spaces are expressed in pericytes and smooth muscle cells.
- Cerebral small vessel disease has almost no genetic overlap with Alzheimer’s.
AD risk genes were mostly expressed in microglia, as expected, with a few in endothelial cells. SVD risk gene expression was scattered across vascular and perivascular cell types, including endothelial cells, pericytes, smooth muscle cells, and astrocytes. Though both diseases disrupted endothelial cell biology, AD and SVD genes hit different processes within that biology. The former predominated in amyloid and lipid pathways, the latter in chromatin regulation and senescence.
Andrew Yang at the University of California, San Francisco, believes that by providing an “epigenomic map” of the main brain and vascular cell types, the data could help pinpoint specific genetic variants that cause disease. “This is an incredibly important paper that creates a foundational resource for the field,” he wrote (comment below).
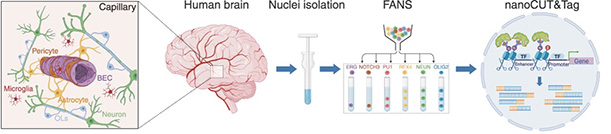
Neurovascular Profiles. After isolating nuclei from brain endothelial cells (BEC) and other cells associated with blood vessels (left) in human brain, scientists separated each cell type by marker genes (middle) and detected regions of open chromatin using antibodies (right). [Courtesy of Ziegler et al., 2025, Neuron.]
Cerebral small vessel disease is an umbrella term for multiple cerebrovascular pathologies, such as hardening or thickening of vessel walls, cerebral amyloid angiopathy, and inflammation around blood vessels. Physicians detect SVD on MRI scans by looking for signs of vessel damage, such as white-matter hyperintensities (WMH), microhemorrhages, lacunar infarcts, and enlarged perivascular spaces. Geneticists have identified more than two dozen genes associated with SVD by GWAS; they have also shown that WMH correlate with a higher risk of AD, linking the two diseases (Sargurupremraj et al., 2020; Sargurupremraj et al., 2024).
To dig deeper into the genetics, Nott and colleagues turned to both biopsy and postmortem tissue. They used cortical epilepsy resections from seven children or young adults, as well as postmortem prefrontal cortex samples from six adults around 80 years old who had been cognitively healthy when they died. Joint first authors Kevin Ziegler and Aydan Askarova isolated nuclei from these samples using fluorescence-activated sorting of six cell types: endothelial cells, mural cells, astrocytes, microglia, oligodendrocytes, and neurons. Mural cells comprise the pericytes and smooth muscle cells that surround capillaries and arterioles, respectively.
The authors identified regions of active chromatin in these nuclei with antibodies against specific methylations and acetylations (image above). This defined the epigenome of each cell type. Next, they looked for noncoding GWAS variants in those cell-specific open chromatin regions to discern which cell types were affected by each disease. Not only was the profile distinct for each disease, but there was almost no overlap. In other words, CSV and AD risk variants were, for the most part, active in different cell types.
Notably, the genetics of SVD depended on which of its phenotypes the authors examined. For WMH, risk genes predominated in astrocytes, with some popping up in vascular cells as well. For enlarged perivascular spaces, risk was concentrated in mural cells. Because perivascular spaces typically expand before WMH form, mural cells might be the site of the earliest changes in SVD, the authors noted.
Given that astrocytes play such a prominent role in SVD, the authors investigated whether these cells contributed to the risk of other cardiovascular conditions such as high blood pressure, stroke, and coronary artery disease as well. Curiously, they found no link. Astrocytes are uniquely associated with SVD, they concluded.
Costantino Iadecola at Weill Cornell Medical College, New York, found this surprising, because hypertension causes SVD. The new data imply that SVD can also be triggered by factors in the brain, independent of blood pressure, he told Alzforum. In a similar vein, Iadecola recently reported that the blood pressure-regulating hormone angiotensin II damages the brain vasculature in mice via mechanisms unrelated to vascular pressure (Nov 2025 news).
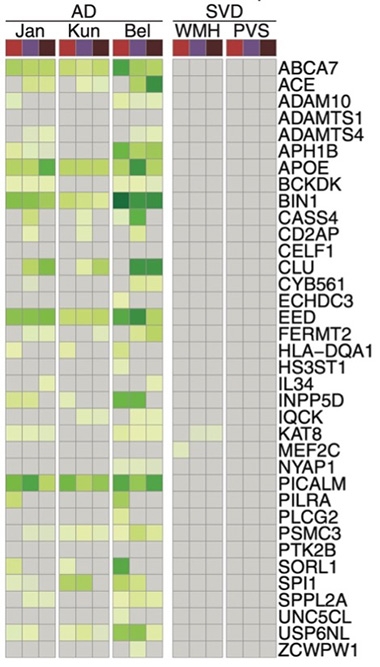
Nothing in Common. AD risk genes (listed on right) identified in three previous GWAS studies (left columns) are expressed in microglia (red), brain endothelial cells (violet), and mural cells (plum). Only two of these 36 genes also associate with small vessel disease (right columns).
Next, Nott and colleagues linked active enhancers and promoters containing GWAS variants to specific genes they influence. For this, they deployed PLAC-Seq, aka proximity ligation-assisted chromatin immunoprecipitation sequencing (Yu et al., 2021). For AD variants, target genes were mainly involved in amyloidosis, lipid metabolism, tau binding, and endocytosis. For SVD, genes were centered in epigenetic modifications, DNA damage, telomere stress, and cellular senescence. In other words, different cellular mechanisms drive AD and SVD (image at right).
Yang recently published a similar study examining how risk variants for multiple neurodegenerative and cerebrovascular conditions alter gene expression in various brain cell types. His team found that neurodegenerative risk genes were concentrated in immune pathways; vascular ones, in extracellular matrix networks (Aug 2025 news). Nott’s findings are consistent with his, Yang noted.
Finally, the authors used published single-cell RNA-Seq datasets to determine if the expression of risk genes predicted by this study did in fact go up or down in microglia in AD brain (Sun et al., 2023; Mathys et al., 2023). They found 91 that did. Then they consulted the L1000 connectivity map, which links drugs to gene expression, to pick out pharmaceuticals that nudge expression in the opposite direction (Subramanian et al., 2017). For example, a vitamin D receptor agonist normalized AD risk gene expression, as did MTOR and HDAC inhibitors. These drugs could be tested therapeutically, the authors suggested. Scientists have tied vitamin D receptor signaling to anti-inflammatory properties of microglia (Aug 2024 conference news).
Christopher Glass at the University of California, San Diego, in whose lab Nott was a postdoctoral fellow, said the next step will be to test these associations in cell culture. “These findings make a strong case for further improving human iPSC-derived models of brain vasculature, so that predictions arising from these studies can be validated with functional assays,” he wrote (comment below).—Madolyn Bowman Rogers
References
News Citations
- Hypertension Hurts Neurons, Oligodendrocytes Within Days
- New Multiomic Atlas to Decode the Brain’s Blood Vessels
- Microglial Epigenetics Hints at How ApoE Toggles Alzheimer’s Risk
Paper Citations
- Sargurupremraj M, Suzuki H, Jian X, Sarnowski C, Evans TE, Bis JC, Eiriksdottir G, Sakaue S, Terzikhan N, Habes M, Zhao W, Armstrong NJ, Hofer E, Yanek LR, Hagenaars SP, Kumar RB, van den Akker EB, McWhirter RE, Trompet S, Mishra A, Saba Y, Satizabal CL, Beaudet G, Petit L, Tsuchida A, Zago L, Schilling S, Sigurdsson S, Gottesman RF, Lewis CE, Aggarwal NT, Lopez OL, Smith JA, Valdés Hernández MC, van der Grond J, Wright MJ, Knol MJ, Dörr M, Thomson RJ, Bordes C, Le Grand Q, Duperron MG, Smith AV, Knopman DS, Schreiner PJ, Evans DA, Rotter JI, Beiser AS, Maniega SM, Beekman M, Trollor J, Stott DJ, Vernooij MW, Wittfeld K, Niessen WJ, Soumaré A, Boerwinkle E, Sidney S, Turner ST, Davies G, Thalamuthu A, Völker U, van Buchem MA, Bryan RN, Dupuis J, Bastin ME, Ames D, Teumer A, Amouyel P, Kwok JB, Bülow R, Deary IJ, Schofield PR, Brodaty H, Jiang J, Tabara Y, Setoh K, Miyamoto S, Yoshida K, Nagata M, Kamatani Y, Matsuda F, Psaty BM, Bennett DA, De Jager PL, Mosley TH, Sachdev PS, Schmidt R, Warren HR, Evangelou E, Trégouët DA, International Network against Thrombosis (INVENT) Consortium, International Headache Genomics Consortium (IHGC), Ikram MA, Wen W, DeCarli C, Srikanth VK, Jukema JW, Slagboom EP, Kardia SL, Okada Y, Mazoyer B, Wardlaw JM, Nyquist PA, Mather KA, Grabe HJ, Schmidt H, Van Duijn CM, Gudnason V, Longstreth WT Jr, Launer LJ, Lathrop M, Seshadri S, Tzourio C, Adams HH, Matthews PM, Fornage M, Debette S. Cerebral small vessel disease genomics and its implications across the lifespan. Nat Commun. 2020 Dec 8;11(1):6285. PubMed.
- Sargurupremraj M, Soumaré A, Bis JC, Surakka I, Jürgenson T, Joly P, Knol MJ, Wang R, Yang Q, Satizabal CL, Gudjonsson A, Mishra A, Bouteloup V, Phuah CL, van Duijn CM, Cruchaga C, Dufouil C, Chêne G, Lopez OL, Psaty BM, Tzourio C, Amouyel P, Adams HH, Jacqmin-Gadda H, Ikram MA, Gudnason V, Milani L, Winsvold BS, Hveem K, Matthews PM, Longstreth WT, Seshadri S, Launer LJ, Debette S. Genetic Complexities of Cerebral Small Vessel Disease, Blood Pressure, and Dementia. JAMA Netw Open. 2024 May 1;7(5):e2412824. PubMed.
- Yu M, Juric I, Abnousi A, Hu M, Ren B. Proximity Ligation-Assisted ChIP-Seq (PLAC-Seq). Methods Mol Biol. 2021;2351:181-199. PubMed.
- Sun N, Victor MB, Park YP, Xiong X, Scannail AN, Leary N, Prosper S, Viswanathan S, Luna X, Boix CA, James BT, Tanigawa Y, Galani K, Mathys H, Jiang X, Ng AP, Bennett DA, Tsai LH, Kellis M. Human microglial state dynamics in Alzheimer's disease progression. Cell. 2023 Sep 28;186(20):4386-4403.e29. PubMed.
- Mathys H, Peng Z, Boix CA, Victor MB, Leary N, Babu S, Abdelhady G, Jiang X, Ng AP, Ghafari K, Kunisky AK, Mantero J, Galani K, Lohia VN, Fortier GE, Lotfi Y, Ivey J, Brown HP, Patel PR, Chakraborty N, Beaudway JI, Imhoff EJ, Keeler CF, McChesney MM, Patel HH, Patel SP, Thai MT, Bennett DA, Kellis M, Tsai LH. Single-cell atlas reveals correlates of high cognitive function, dementia, and resilience to Alzheimer's disease pathology. Cell. 2023 Sep 28;186(20):4365-4385.e27. PubMed.
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, Lahr DL, Hirschman JE, Liu Z, Donahue M, Julian B, Khan M, Wadden D, Smith IC, Lam D, Liberzon A, Toder C, Bagul M, Orzechowski M, Enache OM, Piccioni F, Johnson SA, Lyons NJ, Berger AH, Shamji AF, Brooks AN, Vrcic A, Flynn C, Rosains J, Takeda DY, Hu R, Davison D, Lamb J, Ardlie K, Hogstrom L, Greenside P, Gray NS, Clemons PA, Silver S, Wu X, Zhao WN, Read-Button W, Wu X, Haggarty SJ, Ronco LV, Boehm JS, Schreiber SL, Doench JG, Bittker JA, Root DE, Wong B, Golub TR. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell. 2017 Nov 30;171(6):1437-1452.e17. PubMed.
Further Reading
Primary Papers
- Ziegler KC, Askarova A, Gergian C, Yaa RM, van Dalen JD, Transfeld JL, Zoppi F, Du J, Clode D, Rahbar F, Graham RE, Sargurupremraj M, Debette S, Gonda DD, Levy ML, Chandran S, Falk S, Karow M, Matthews PM, Coufal NG, Nott A. The brain neurovascular epigenome and its association with dementia. Neuron. 2025 Oct 30; Epub 2025 Oct 30 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
UCSF
I think this is an incredibly important and exciting paper and creates a foundational resource for the field. It provides high-resolution epigenomic maps—essentially the gene regulatory blueprint—for purified human brain vascular cells (endothelial and mural cells) and other key brain cell types. This allows us to interpret large-scale genetic studies and pinpoint where the risk for these diseases actually functions.
A key advance is the robust cell type-specific partitioning of genetic risk. The data confirm that AD heritability is overwhelmingly driven by microglia (the brain's immune cells), and brain endothelial cells. In contrast, cerebral small vessel disease (CSVD) heritability is spread across the entire neurovascular unit (NVU), including astrocytes. The study also revealed minimal overlap of vascular and immune putative risk genes in AD and SVD. This is consistent with our own recent findings, where we observed that AD variants dysregulate inflammatory adaptor proteins in vascular and immune cells, while cerebrovascular disease variants disrupt extracellular matrix genes crucial for vessel integrity (Reid et al., Neuron 2025).
From the perspective of inherited genetic risk, the data strongly indicate that the underlying genetic drivers of AD and CSVD are largely distinct (again, consistent with our own recent findings). I think that is perhaps the most striking finding. Even when the same cell type, e.g., brain endothelial cells, is involved in both diseases, the biological pathways affected are different. The authors found that AD risk variants point towards amyloid processing and lipid pathways, whereas CSVD variants point strongly towards cellular senescence and chromatin regulation.
However, I think it is also important to distinguish between genetic origin and pathological interaction. While the genetic roots of AD and CSVD appear distinct, these diseases clearly interact in the brain once they manifest, likely exacerbating cognitive decline in the large majority of AD patients who have mixed pathology.
To me, the most exciting next step is functional validation and mechanistic understanding of these distinct pathways. For example, modeling these variants in cell lines or animal models to understand how they work, how they interact with pathologies, and how one may target them therapeutically. It will also be valuable to expand on this important study to larger and more diverse sample cohorts. Finally, this study analyzes bulk sorted cell populations—with single-cell technologies, it will be exciting to analyze how disease genetic risk manifests not just in specific cell types, but in specific disease-associated cell states (e.g., reactive astrocytes or T cell subsets).
Rush University Medical Center
This work is interesting because the authors quantified epigenomic changes in the promoter and enhancer regions of human cortical brain cells, correlating with risk of AD, small vessel disease (SVD), and other vascular related traits, such as systolic and diastolic blood pressure and atrial fibrillation. They used publicly available data. Consistent with prior studies, they found AD genetic risk variants significantly enriched in the microglial regulatory genome while SVD risk variants were enriched in neurovascular unit cells, such as endothelial cells, vascular mural cells (vascular smooth muscle cells and pericytes), and astrocytes. However, there was modest enrichment of AD risk variants in the neurovascular unit cells too, including endothelial cells and vascular mural cells. The latter was suggestive of the biological basis for the association between AD and SVDs, such as CAA.
However, more thorough examination of the genes affected by the disease-correlated promoters/enhancers in different cell types indicated that AD-related microglial and endothelial genes were enriched in pathways (such as amyloid-beta formation) different than SVD-related microglia and endothelial cells genes (such as senescence-associated secretory phenotype). This indicates that AD and SVD have different underlying molecular pathways.
The authors used in silico techniques to predict AD and SVD treatments based on the genes whose expression was perturbed by genetic variants in the promotor/enhancer regions of different cell types. However, this may be too ambitious because it is unclear if the expression of those genes’ will change AD or SVD in the real world.
The study confirms prior studies’ findings about some of AD risk genes and suggests new risk variants and genes. Highlighting endothelial and vascular cells beyond microglia as the cells with AD genetic risk variants provide cellular basis for the studies that report vascular contributing to AD pathophysiology. The main limitation of the study is the small sample size (n=13) of the brains whose cortical cells’ epigenetic data were quantified, and using summary data rather than person-level data for inferring most of the study findings.”
University of California San Diego
I am excited to see this new paper from Alexi Nott, in which he and his lab significantly extend his prior studies of epigenetic landscapes to now include brain endothelial cells and mural cells. By comparing the active regulatory regions of these cells with those of previously studied brain cell types, he confirms and extends the enrichment of non-coding risk alleles for AD in microglia enhancers. In addition, he now shows that risk alleles for cerebral small vessel disease are enriched in brain endothelial cells, mural cells, and astrocytes, implicating actions of the linked genes to functions of the neurovascular unit.
In addition to being an important new epigenetic resource, these findings make a strong case for further improving human iPSC-derived models of brain vasculature so that predictions arising from these studies can be validated with functional assays.
University College London
Ziegler, Askarova, and colleagues investigated AD and SVD genetic risk associated with gene regulatory landscapes of human brain vascular cells. Their key findings show that AD genetic risk predominantly affects microglia, which is consistent with previous studies [e.g., Sudwarts & Thinakaran, 2023; Graham et al., 2025], with minimal involvement of endothelial cells. Notably, the study broadens our understanding of genetically driven cell type-specific risk in SVD, implicating a wider range of brain cell types, including astrocytes.
A particularly important aspect of the study is the introduction of a genetics-based framework for drug prioritization, which complements other recent approaches using multi-omics in the AD field [e.g., Lui et al., 2025].
The analysis of endothelial enhancer-to-gene interactomes identified genes associated with amyloid processing and immune pathways in AD, and with chromatin regulation and neurovascular cell senescence in SVD, highlighting distinct molecular pathways in these frequently co-occurring diseases. Conversely, transcription factor motif analysis revealed potential shared mechanisms between AD and SVD, such as alterations in the type I interferon response, suggesting convergent pathogenic processes influenced by disease-associated variants. This work contributes valuable insights into the overlapping and divergent molecular underpinnings of AD and SVD. Yet, does it complete the puzzle?
Although oligodendrocytes have previously been implicated in AD genetic risk [e.g., Graham et al., 2025], and their interactions with endothelial cells are known to be critical in SVD [Rajani & Williams, 2017], this study did not detect significant genetic susceptibility within oligodendrocytes for either condition. Given that demyelination occurs early in both AD and SVD, likely due to oligodendrocyte dysfunction and their inability to regenerate myelin [e.g., Fodder et al., 2023], this absence raises intriguing questions. Could oligodendrocytes be less affected by the epigenetic marks examined in this study? Supporting this hypothesis, our recent research points to abnormal DNA methylation as a key driver of oligodendrocyte dysfunction in AD and potentially in other neurodegenerative diseases [Fodder et al., 2025].
Moreover, could cellular heterogeneity, not captured by the FANS approach used here, obscure additional signals? Could disease context or age-related changes disrupt the epigenetic landscapes described here? Integrating single-cell methodologies, more comprehensive multi-omics, multimodal data frameworks, and functional validation in appropriate models, will be essential to unravel the full complexity of AD and SVD.
Overall, this is an exciting study that brings new insights and testable hypotheses to the field.
King's College London
King's College London
The continuous development and refinement of cell-type–specific and single-cell methodologies have highlighted the importance of understanding cellular heterogeneity in neurodegeneration by revealing distinct (epi)genetic signatures and underlying gene regulatory mechanisms. Observationally, AD patients frequently exhibit concomitant vascular pathology, including Cerebral Amyloid Angiopathy, a key feature of small vessel disease. However, understanding how vascular and perivascular cells contribute to the pathogenesis of neurodegenerative diseases, such as Alzheimer’s and SVD, and whether genetically implicated disease mechanisms converge has been challenging. The inherent scarcity and fragility of these cell types pose considerable experimental challenges, hindering efforts to investigate neurovascular epigenetic and gene regulatory mechanisms.
Novel approaches like VINE-Seq (Vessel Isolation and Nuclei Extraction) and, more recently, MultiVINE-Seq have advanced these efforts. MultiVINE-Seq captures paired transcriptomic and epigenomic data from human brain vascular, perivascular, and immune cells via single nucleus RNA (snRNA-seq) and Assay for Transposase-Accessible Chromatin (snATAC-Seq) sequencing, respectively (Reid et al., 2025). While MultiVINE-Seq has enabled the profiling of accessible chromatin, the comprehensive enhancer and chromatin interaction landscape of neurovascular cells and their association with genetic risk have remained largely unexplored.
In this study, Ziegler, Askarova, and colleagues report histone acetylation (H3K27ac) and methylation (H3K4me) profiles, together with chromatin interactions, of six brain cell types, including cells of the neurovascular unit (NVU), which comprise brain endothelial cells (BECs), mural cells, astrocytes, and microglia. H3K27ac, enriched at active enhancers and promoters, and H3K4me3, marking active promoters, were profiled via an adapted nanobody-based cleavage under targets and tagmentation (nano-CUT&Tag) assay, while Proximity ligation-assisted ChIP-Seq (PLAC-Seq) and promoter capture Hi-C were used to map chromatin contacts across the genome in these cells.
A first major result of this study is that AD heritability is consistently associated with myeloid/microglia enhancers, reinforcing prior links to immune regulatory regions, with a modest enrichment in BECs. On the other hand, SVD heritability was enriched across the neurovascular unit, specifically BECs, mural cells and astrocytes. Employing Hi-C-coupled Multimarket Analysis of Genomic Annotation (H-MAGMA) the authors identified distinct putative AD and SVD risk genes and pathways in microglia and BECs. While AD risk genes were involved in Aβ formation and processing, phospholipid and protein-lipid complex, tau protein binding, and endocytosis, SVD risk genes were associated with chromatin regulation, senescence, telomere stress, and DNA damage.
Additionally, the authors found that 78 H-MAGMA-identified risk genes for AD in microglia were also differentially expressed in AD. They leveraged this finding to nominate two putative repurposable drugs, BRD-K63513868 and calcitriol, that could potentially reverse the AD-associated expression patterns of these risk genes. Interestingly, calcitriol is a vitamin D receptor (VDR) agonist, and our lab has recently reported increased VDR binding in APOE2 microglia xenotransplantated into the brains of APPNL-G-F mice. Our work suggests that VDR activation may contribute to the isoform’s protective role in AD via enhancing anti-inflammatory signalling (Murphy et al., 2025). This study by Ziegler, Askarova and colleagues provides further evidence that VDR activation may exert protective effects on microglia in the context of AD.
Moreover, this study examined how AD and SVD variants alter transcription factor (TF) motif binding at PU1 and ERG regions, lineage-determining TFs for myeloid/microglia and endothelial cells, respectively. Importantly, a variety of motifs altered by AD variants were found on chromosome 19 at the APOE locus, implicating APOE itself and corroborating AD-associated regulatory changes linked to APOE in microglia explored by our previous work.
Beyond providing critical insights into epigenetic signatures and associated gene regulatory mechanisms of neurovascular and brain parenchymal cell populations in AD and SVD, this study also serves as a valuable framework for further research efforts to functionally dissect cell-type-specific mechanisms and explore therapeutic avenues in neurodegenerative diseases.
References:
Reid MM, Menon S, Liu H, Zhou H, Hu Z, Frerich S, Ding B, Oveisgharan S, Zhang Z, Nelson S, Apolonio A, Bennett DA, Dichgans M, Pollard KS, Corces MR, Yang AC. Human brain vascular multi-omics elucidates disease-risk associations. Neuron. 2025 Jul 23; Epub 2025 Jul 23 PubMed.
Murphy KB, Hu D, Wolfs L, Rohde SK, Fauró GL, Geric I, Mancuso R, De Strooper B, Marzi SJ. The APOE isoforms differentially shape the transcriptomic and epigenomic landscapes of human microglia xenografted into a mouse model of Alzheimer's disease. Nat Commun. 2025 May 27;16(1):4883. PubMed.
Make a Comment
To make a comment you must login or register.