CONFERENCE COVERAGE SERIES
LATE (Limbic-predominant Age-related TDP-43 Encephalopathy) 2022
Online Meeting
11 February 2022

CONFERENCE COVERAGE SERIES
Online Meeting
11 February 2022
A virtual workshop on limbic predominant age-related TDP-43 encephalopathy, held virtually February 11, began with a moment of silence for the late John Trojanowski. His work played a critical role in nabbing TDP-43 inclusions as a culprit behind frontotemporal lobar degeneration and amyotrophic lateral sclerosis, and later, he helped peg the same RNA-binding protein as the critical component in what is now known as LATE—a vastly more common neuropathological entity that comes with memory loss.
At the workshop, researchers joined from across the aisles of FTD/ALS and AD research to carve out a niche for LATE, which was christened as a distinct neuropathological disease only three years ago. Since then, researchers have painstakingly plumbed brain samples from multiple autopsy cohorts, further refining the neuropathological characteristics of LATE-neuropathological change (NC), its prevalence, and how this flavor of TDP-43 pathology contributes to cognitive decline, particularly in comparison to its more famous amnestic doppelganger, AD.
As researchers at the meeting presented these findings from cohort after cohort, it became clear that LATE-NC is common, cropping up in around a third of brain samples from people who died in their 80s and 90s. Several investigators reported that this limbic pathology contributes substantially to cognitive decline, either by itself or in combination with AD, casting LATE as a public health threat in its own right. It may also be a meddler in clinical trials for AD, researchers lamented, because it can accelerate that dementia. As of now, a LATE diagnosis can only be made at autopsy, and biomarkers are sorely needed to detect it during life. A few contenders—including fluid and neuroimaging biomarkers—ended the meeting on a promising note.
Co-chaired by Peter Nelson of the University of Kentucky in Lexington and Julie Schneider of Rush University in Chicago, the five-hour meeting hosted by the NIA drew more than 350 attendees from 14 different countries. It featured 19 talks on the neuropathological, clinical, epidemiological, and genetic aspects of LATE, along with 25 prerecorded poster narrations. The workshop convened researchers from different “cliques” in the dementia field, Nelson said. This was by design. “To have all these factions talking to each other, and listening to each other, was very productive,” Nelson told Alzforum.
Others agreed. “It was important to bring different people together to really define the urgent questions that need to be resolved about LATE,” said Manuela Neumann of the German Center for Neurodegenerative Diseases in Tübingen.
“I was pretty blown away by the whole meeting,” said Robert Rissman of the University of California, San Diego, who presented preliminary findings on a new biomarker for TDP-43. “There’s a lot of excitement in this field right now, but still so much we don’t know,” Rissman said.
TDP-43 arrived on the scene in 2006, when Neumann, then a visiting scholar in Trojanowski’s lab at the University of Pennsylvania, Philadelphia, identified ubiquitinated inclusions of the protein as the hallmark pathology in many cases of FTLD and ALS (Oct 2006 news; Cairns et al., 2007). Soon after, neuropathologists started detecting TDP-43 inclusions in people with AD and/or hippocampal sclerosis, which causes severe neuronal loss and gliosis in the hippocampus (Amador-Ortiz et al., 2007). In these cases, which tended to have AD-like symptoms rather than symptoms of FTLD or ALS, TDP-43 inclusions predominated in the limbic regions, including the amygdala and hippocampus.
Not all cases of this limbic TDP-43 pathology come with hippocampal sclerosis, but vice versa, researchers have found that the vast majority of hippocampal sclerosis is accompanied by limbic TDP-43. In recognition of this pattern, a cadre of researchers held a workshop in 2018 to forge a consensus on the nature of the beast. From that meeting, “LATE” was born, along with a proposed neuropathological staging scheme (May 2019 news). Based primarily on autopsy data from the Religious Orders Study (ROS-MAP), researchers proposed that TDP-43 inclusions appear in the amygdala in stage 1, then the hippocampus and entorhinal cortex in stage 2, and finally the neocortex in stage 3 (James et al., 2016). Cognitive impairment tended to start once the pathology spread beyond the amygdala. As its name suggests, LATE arises late in life, lurking in the brains of 20 percent of people in their 80s who had received a dementia diagnosis, at least in the ROS-MAP cohort.
At the time, not everyone was jazzed about the new moniker. Some researchers questioned the need to name a new disease after what was primarily a neuropathological phenomenon with uncertain clinical effect (Josephs et al., 2019). They also called for further work to distinguish LATE-NC from FTLD.
In response to the latter challenge, Nelson led a study that tasked neuropathologists with discriminating between FTLD-TDP-43 and stage 3 cases of LATE-NC from autopsy cohorts at the University of Kentucky and the University of Pennsylvania (Robinson et al., 2020). Using prespecified criteria based on where and how severe TDP-43 pathology was in the brain—limbic areas for LATE and frontal cortex for FTD—the pathologists, who were blind to the diagnosis of the patients, correctly distinguished between the two disorders more than 90 percent of the time. When cases with less severe stages of LATE-NC were included in the analysis, overlap of regional pathology between the two diseases shrank. The specificity of telling the two apart rose to 98 percent, because TDP-43 pathology in earlier stages of LATE-NC is more tightly confined to limbic regions, clearly distinguishing it from FTLD.
Better Staging
At the meeting, Alexandra Young of King’s College London described a staging scheme based on SuStain, a machine-learning analysis that uses neuropathological data from hundreds of brain samples to identify patterns of pathology progression (Oct 2018 news). Young previously used SuStain to identify different subtypes of AD (Apr 2021 news). Applying the algorithm to brain samples from the University of Pennsylvania brain bank, Young identified stages of LATE-NC that roughly aligned with previously proposed staging schemes, with TDP-43 aggregates starting in the amygdala, moving into the hippocampus and entorhinal cortex, and finally out into the neocortical regions. The model also distinguished between LATE-NC, FTLD-TDP, and ALS patterns of neuropathology 96 percent of the time. This was regardless of whether people with LATE-NC also had AD. “That’s a better diagnostic specificity than we can achieve with many dementia pathologies,” Nelson said during his presentation at the meeting, referring to both his published findings and Young’s new data.
The difference between most cases of FTLD and LATE-NC might appear obvious to the discerning eye of a neuropathologist, or a computer algorithm; even so, the official neuropathological criteria for LATE-NC need to be updated to reflect these distinctions, Neumann emphasized in her talk at the meeting. Specifically, she noted that based on the current criteria, all cases of FTLD-TDP-43 and 30 to 60 percent of ALS cases, in which TDP-43 inclusions are found in the frontal cortex, could technically be classified as stage 3 LATE-NC.
Neumann thinks that clearly defined exclusion criteria are needed. For example, if TDP-43 inclusions crowd the primary motor cortex, as in ALS, that would eliminate LATE-NC. Similarly, if cortical inclusions are not Type A—defined as small, compact, cytoplasmic, and in the upper layers of the cortex—then LATE-NC should be ruled out. That is because so far, only Type A inclusions have been detected in cases of LATE-NC, whereas any of five morphological subtypes of TDP-43 pathology—types A to E—can be found in cases of FTLD. “If it’s not Type A, I would call it FTLD-TDP,” she said. When cases are difficult to call, Neumann noted that antibodies specific for different subtypes of TDP-43 pathology could come in handy. At the workshop, she showed preliminary data from two such antibodies—one that bound only Type A TDP-43, and one that bound all other types.
Clinical Relevance
Neumann also thinks that LATE-NC diagnoses need to be clinically relevant. As of now, a single TDP-43 inclusion in the amygdala would be grounds for diagnosing stage 1 LATE-NC. In a person with dementia, this scant level of pathology could not account for their decline. This problem harkens back to the early days of AD neuropathology research, Neumann said, when some might have considered a single amyloid plaque a sign of the disease. Neumann believes the staging criteria are too simplistic, and that the field needs more quantitative specificity that further defines both the neuropathological stages and their contributions to cognitive symptoms.
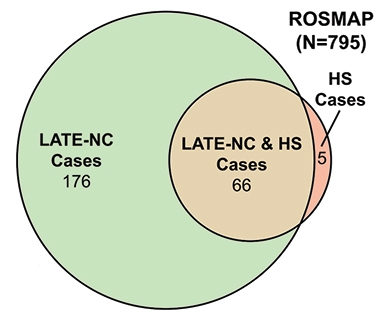
HS and LATE-NC. The vast majority of HS cases occurred in the context of LATE-NC, shown here in the ROS-MAP cohort. [Courtesy of Dugan et al., Neurology, 2021]
Nelson and others agreed that such updates to the LATE-NC criteria will be crucial moving forward, especially as more neuropathologists become familiar with this new category.
Overlap with TDP-43 pathology in other diseases has complicated the interpretation of LATE. For example, since the 2019 consensus report, scientists have delved into the relationship between LATE-NC and hippocampal sclerosis. Although the latter is commonly seen along with LATE-NC, it is not tied to any particular disease, and has been seen in people with other neurodegenerative diseases including AD and FTLD, as well as in the wake of neurological insults such as epilepsy and stroke. One study examining samples of more than 1,400 brains from the National Alzheimer’s Coordinating Center (NACC) and ROS-MAP cohorts, found that 66 of 77 cases of HS also had LATE-NC (Dugan et al., 2021; see image above). Another study examined samples from more than 400 brains in the NACC, finding that among 366 people with LATE-NC, 145 who also had hippocampal sclerosis trended toward having been more cognitively impaired during life, and tended to have more widespread TDP-43 pathology (Gauthreaux et al., 2022). The findings suggest that hippocampal sclerosis may represent a more advanced stage of LATE-NC, although the exact mechanisms linking the two remain unclear, Nelson said.
How Common is LATE?
Nelson presented fresh findings from the largest study on this question done to date. Because most research and clinic-based autopsy cohorts skew toward people with neurodegenerative disease, Nelson sought samples that were more representative of the general population. In all, he compiled autopsy data from 13 community- or population-based cohorts, comprising samples from 6,251 brains. Participants were an average of 88 years old when they died, and a quarter were ApoE4 carriers. Nelson reported that 39 percent of them had LATE-NC. Of those, a third were in LATE-NC stage 1, with TDP-43 pathology confined to the amygdala region. About a quarter of people across these cohorts had stage 2 or 3 LATE-NC, suggesting it could contribute to cognitive impairment.

LATE-NC Without AD. Among people who had no substantial AD pathology, more than a quarter had LATE-NC. [Courtesy of Peter Nelson, University of Kentucky.]
Nelson also examined relationships between AD pathology—in this case, the CERAD neuritic amyloid plaque score—and LATE-NC. Two-thirds of cases had CERAD scores indicative of at least some AD pathology, and about half of these also had LATE-NC. Of the one-third of participants whose low CERAD scores indicated little to no AD pathology, 27 percent had LATE-NC. The findings suggest that while LATE-NC is most prevalent among people with AD pathology, it also occurs on its own (see image below).
Nelson pointed out that, so far, studies suggest that LATE-NC on its own packs less of a wallop on cognition than does AD, resulting in a slower course of decline (Boyle et al., 2017; Nag et al., 2017). However, he noted that pure LATE-NC and pure AD are uncommon—far more often, both pathologies accumulate in a person's brain, resulting in a faster course of disease than either alone. Nelson said that biomarkers are desperately needed to diagnose LATE-NC, to study its natural history and role in cognitive decline, and to better plan and test outcomes in clinical trials (see Part 2 of this series).—Jessica Shugart
No Available Further Reading
Scientists increasingly recognize limbic predominant age-related TDP-43 encephalopathy—aka, LATE—as a highly prevalent neuropathology lurking in the brains of people in their 80s and 90s. While neuropathologists have long observed TDP-43 inclusions crowding the limbic regions among people with memory complaints, the christening of this neuropathological entity nearly three years ago has prompted them to re-examine autopsy cohorts, and decipher how much this pathology contributes to cognitive impairment. At LATE 2022—a virtual workshop hosted by the National Institute on Aging on February 11—researchers reported that not only was LATE neuropathology highly prevalent in older people, but it also mattered. People who had LATE pathology at death were likelier to have suffered from cognitive impairment or dementia during life, and they had a similar, albeit less severe, amnestic syndrome as that caused by AD. When lurking together in the same brain, LATE and AD pathology dramatically stepped up risk for dementia, suggesting a synergism between these neuropathological scourges of aging.
LATE Decline
As people age into their 80s and 90s, mixed proteopathy increasingly becomes the name of the game and brains dotted with pure neuropathologies become less common. LATE arises during this period of aging. At the NIA workshop, researchers questioned how much LATE neuropathological change (LATE-NC) contributes to cognitive decline, especially if it occurs in the company of other protein deposits.
Patricia Boyle of Rush University Medical Center in Chicago addressed this in the Religious Orders and Memory and Aging Project (ROS-MAP), a community cohort of some 3,000 people who had no dementia at baseline and submitted to lifelong cognitive testing and an autopsy after death. Focusing on participants who had undergone at least three cognitive examinations, Boyle asked to what extent different pathologies had contributed to a person’s deviation from the average pace of cognitive decline. She noted that in past studies, a combination of 11 brain pathologies, including amyloid plaques, neurofibrillary tangles, α-synuclein aggregates, limbic TDP-43 inclusions, hippocampal sclerosis, and various vascular pathologies accounted for 43 percent of that variance (Boyle et al., 2021). Myriad other factors, including lifestyle, cognitive resilience, education, and socioeconomic status, also steer cognitive trajectories (Jan 2017 news; Oct 2018 news; Oct 2019 news; Jan 2019 news).
In her previous study, Boyle had assumed a linear course of cognitive decline. At LATE2022, she presented findings from an updated statistical model, called functional mixed effects, that accounts for ups and downs of cognitive decline. Boyle said that flexibility built into this model allows it to more closely track how a person’s cognitive function fades. Among 1,147 ROS-MAP participants, global AD pathology accelerated decline by 26 percent, TDP-43 pathology by 8.3 percent, and hippocampal sclerosis by 6.6 percent. Cerebral amyloid angiopathy explained 5.7 percent of the change, while all other vascular pathologies combined explained less than 3 percent. Boyle found that, in toto, the measured pathologies accounted for half the cohort’s variance in cognitive decline. She said this finding implicates LATE-NC as a driver of late-life cognitive decline.
To be clear, this is not to say that half of ROS-MAP participants lacked a neuropathological explanation for their cognitive impairment. Rather, the remaining variance could stem from myriad nonpathological factors or unknown pathologies that were not measured by the study, said co-author Julie Schneider from Rush.
Do LATE-NC and AD harm the mind in the same way? Boyle investigated this by examining individual cognitive domains. Both LATE-NC and AD associated with a dip in episodic memory about 10 years prior to death, and with semantic memory loss appearing five years later. Working memory started to falter a few years earlier in people with plaques and tangles than in people with LATE-NC, and visuospatial abilities were more severely degraded in cases with AD pathology than LATE-NC.
“In terms of cognitive domains, there are more similarities than differences between LATE-NC and AD,” Nelson said. “Pure AD is more severe than pure LATE-NC, but the two pathologies are more likely to occur in combination than alone,” he added.
Mixed Pathologies
Indeed, Nelson and colleagues previously reported that among 375 participants in the University of Kentucky ADRC autopsy cohort who died at an average age of 87, around 13 percent had not one but four neurodegenerative proteinopathies: amyloid plaques, neurofibrillary tangles, TDP-43 inclusions, and α-synuclein Lewy bodies. This is in keeping with a large number of studies documenting mixed pathology in AD (Karanth et al., 2020; Schneider et al., 2007; James et al., 2016).
Correlating that ADRC neuropath data with cognition, U. Kentucky’s Erin Abner reported that people with those same four misfolded proteinopathies deteriorated faster through mild cognitive impairment and into dementia than did people with fewer proteinopathies. Among those in the cohort who died with dementia, 19 percent had all four, 27 percent had plaques, tangles, and TDP-43 inclusions, while 5 percent had tangles and TDP-43. Notably, all participants included in this analysis had at least some tangle pathology consistent with primary age-related tauopathy. Abner said her findings place TDP-43 among the heavy hitters on cognition in this age bracket.
Yet more support for LATE-NC as a cognitive menace came from the population-based Adult Changes in Thought study. Caitlin Latimer of the University of Washington in Seattle presented preliminary findings from ACT, which enrolls cognitively normal people over the age of 65 in brain imaging, cognitive testing, and, ultimately, neuropathology. ACT researchers recently finished LATE-NC staging across the entire autopsy cohort, which currently includes brain samples from more than 800 people. Latimer investigated how their LATE-NC relates to their plaques/tangles, and dementia. In agreement with findings in other cohorts, Latimer saw both LATE and AD neuropathology as strongly linked to dementia. Compared to 271 people who had neither, 61 people with LATE-NC alone were twice as likely to have died with dementia, while 216 people with AD pathology alone had nearly quadruple the risk. The 127 people with both LATE-NC and AD pathology were 18 times likelier to have had dementia at death. Latimer said her findings were controlled for sex and age, ruling out the possibility that people with LATE-NC were likelier to have had dementia merely because they tended to be older than other members of the cohort.
Latimer noted that nearly everyone with both LATE-NC and AD had died with dementia, suggesting the combination of the two pathological entities potently drives decline. Is this relationship merely additive, Latimer wondered? Her preliminary findings hinted at potential synergism between LATE-NC and tangles, though not amyloid. The presence of stage 2-3 LATE-NC correlated with a higher burden of tangles, particularly in the midfrontal gyrus, Latimer reported, suggesting that neocortical tangles and limbic TDP-43 deliver a one-two punch on cognition.
In the Oldest Old
How does LATE-NC affect cognition in the oldest old? Maria Corrada of the University of California, Irvine, investigated this in the 90+ study, a cohort of nona- and centenarians based in Orange County, California. So far, the study has banked brain samples from 401 people who died at an average of 98 years of age, 44 percent of them with dementia. A little more than a third had had LATE-NC, most of them stage 2. After age 100, the prevalence of LATE-NC rose to 44 percent, from 33 percent in nonagenarians. In contrast to data from other younger cohorts, Corrada found that even stage 1 LATE-NC—in which TDP-43 inclusions are only in the amygdala—boosted the person’s dementia risk. In fact, LATE-NC of any stage more than tripled risk of dementia in this cohort.
There were cases of hippocampal sclerosis in this cohort, as well (see also Part 1 of this series). Two-thirds of them cropped up in people with limbic TDP-43 pathology, supporting the idea that most of this hippocampal pathology is associated with LATE-NC.
Corrada also probed how LATE-NC fits in with the burden of AD pathology, finding that LATE-NC correlated with Braak stage as well as the CERAD score of neuritic amyloid plaques. Although these two pathologies correlated, Corrada emphasized that it was not uncommon to see LATE-NC in people without substantial AD pathology, and that LATE-NC appeared to exert an independent effect on cognition.
The majority of people in this cohort died with more than one type of pathology in their brain, and Corrada used a statistical model to estimate how each may have contributed to dementia. She reported that vascular pathology posed by far the greatest risk, accounting for 40 percent of dementia. AD was next at 25 percent, and LATE-NC, Lewy bodies, and hippocampal sclerosis accounted for 19, 5.5, and 2.7 percent, respectively.
In discussion during the meeting, Carol Brayne, Cambridge University, England, picked up on the apparent differences in how much vascular pathology contributes to dementia across cohorts. While Abner had attributed 3 percent contribution to vascular problems in the University of Kentucky ADRC cohort, Corrada reported that it accounted for 40 percent of dementia risk in the oldest old. Corrada said that in addition to LATE-NC, vascular pathology, in particular small microvascular lesions, contributes substantially to the rising dementia rates as people live to be very old. She lamented that these oldest age groups are excluded from most clinical trials (see Part 3 of this series for more about LATE clinical trials and emerging biomarkers). “We’re neglecting a very high proportion of people who are actually affected by this condition,” she said.—Jessica Shugart
No Available Comments
No Available Further Reading
Limbic predominant age-related TDP-43 encephalopathy, aka LATE, appears to dampen an aging person's memory in a similar way as does Alzheimer’s disease. TDP-43 inclusions occur as the lone pathology in some people, and alongside the plaques and tangles of AD in others. Alas, there are no biomarkers for TDP-43 inclusions. This poses a tricky challenge for the diagnosis and management of both disorders. At LATE 2022, a virtual meeting hosted by National Institute on Aging on February 11, trialists voiced concern that undetected LATE neuropathology in some enrollees of AD drug trials might skew the trial’s outcomes. They also said the field needs trials specifically for LATE.
Researchers described their quest to find biomarkers for LATE. Soluble TDP-43 packaged within astrocytic exosomes detected in blood tracked remarkably well with LATE pathology. PET tracers for the TDP-43 inclusions remain elusive, but other neuroimaging modalities, for example structural change in the hippocampus and patterns of hypometabolism, may help identify people with LATE. Even so, scientist at the meeting agreed that much more work needs to be done to characterize these early leads.
LATE and Trials
Given recent work tying LATE to cognitive decline (see Part 2 of this series), scientists now think it’s quite plausible that this limbic pathology, unbeknownst to trialists, could influence outcomes in AD clinical trials. For example, LATE could mask otherwise positive results of drugs targeting plaques or tangles.
Reisa Sperling, Brigham and Women’s Hospital, Boston, leads secondary prevention trials for AD. She wondered to what extent LATE might explain outliers in those studies—i.e., people who decline much faster than was predicted based on their levels of plaque and tangle pathology at baseline. To get a sense of this, Sperling looked to observational studies, including the Harvard Brain Aging study, ROS-MAP, and ADNI. She noted that while as a group, cognitively normal people with amyloid plaques are likely to slip on cognitive tests in the coming five years, individual trajectories vary dramatically.
Similarly, tau tangles spread at varying rates in the brain. While they eventually invade the neocortex in most people with elevated plaque burden—a scenario Sperling and others call a “cataustrophe,” in some, tangles never make this move, yet the person still slides quickly into cognitive impairment, Sperling noted. Could LATE explain some of these differences?
In the HABS cohort, low hippocampal volume strongly predicted cognitive decline, even among people who had no Aβ deposition, Sperling said, hinting they might indeed have LATE, but without autopsy data or a biomarker, this is but a hunch.
Hints that LATE causes cognitive decline also came from ApoE4 carriers, who are likelier to get AD than noncarriers. While cognition in carriers declined faster than in noncarriers, in some, this happened independently of both plaques and tangles, Sperling said. Since recent studies suggest that ApoE4 boosts risk for LATE, perhaps the risk allele can speed decline via TDP-43 in people with or without AD pathology, Sperling speculated.
In toto, outlier observations such as these offer faint signals that LATE plays some kind of role in determining when cognitively normal people who test positive for amyloid—the population enrolled in secondary prevention trials—slide toward cognitive impairment.
Sans biomarkers, there is no way to directly measure TDP-43 pathology during life, hobbling researchers who want to study how LATE affects cognitive decline, or recruit participants into trials that try to prevent it.
While the biomarker search is on, researchers led by Gregory Jicha, University of Kentucky, Lexington, are relying on the process of elimination to conduct what Peter Nelson, also at U. Kentucky, described as the first-ever treatment trial for LATE (see clinical trial). It recruits people who come to the U. Kentucky ADRC with memory problems yet test negative for amyloid. Those whose total tau is elevated and hippocampal volume reduced—two markers of neurodegeneration—will be invited to enroll. The trial will test nicorandil, a vasodilator developed to treat angina and other heart conditions, as a treatment for hippocampal sclerosis of aging, a pathology that most often co-occurs with LATE (see Part 1 of this series; Nelson et al., 2014; Nelson et al., 2015; Dugan et al., 2021). The drug targets SUR2, a subunit of a potassium channel. This channel senses stress or hypoxia, and helps regulate the neurovascular unit to increase blood flow. It is encoded by ABCC9, a gene that has been identified as a risk factor for hippocampal sclerosis.
“We think that part of what turns LATE into hippocampal sclerosis is a vascular problem,” Nelson told Alzforum. In support of this idea, Nelson said that in people with LATE and hippocampal sclerosis, TDP-43 inclusions are found in glial processes around small blood vessels.
TDP-43 Biomarkers
Where does the search stand for biomarkers to help assess if TDP-43 pathology influences AD trials, and to take direct aim at it with a therapeutic? Robert Rissman of the University of California, San Diego, suggested that TDP-43 packaged inside exosomes could signal the presence of LATE in the brain. Rissman highlighted recent work indicating that misfolded TDP-43 propagates between brain cells in AD, FTD, and ALS (Oct 2018 news; Jo et al., 2020). It does so via several routes, including exosomes. Many different cell types in the brain secrete these tiny membrane packages, and some manage to find their way into the blood, where their neural origins can be deciphered with cell-type-specific markers.
Previous studies have implicated exosomes in the propagation of other proteopathic proteins, including tau and α-synuclein, though the brain origin of plasma exosomes is also dogged by some controversy (Dec 2016 news; Jun 2021 news).
Might TDP-43-laden exosomes serve as a plasma biomarker for LATE? To find out, Rissman and colleagues acquired banked plasma samples from brain donors in the University of Kentucky autopsy cohort, including 22 people with LATE and 42 without. Rissman then used an IgG1 antibody that recognizes TDP-43, regardless of its phosphorylation status, to create a custom assay. Postdoc Charisse Winston sorted exosomes in the plasma by brain-cell-type origin, cracked them open, and looked for TDP-43.
What she found was surprising, Rissman said. While neuronal exosomes contained a substantial, and similar, amount of TDP-43 regardless of whether the donor had LATE pathology, astrocytic exosomes only contained TDP-43 if they came from the blood of a person with LATE. Rissman was shocked at how well the assay differentiated people with or without the pathology, adding that only four people without LATE had any detectable TDP-43 in their astrocyte exosomes, and it was minuscule compared to the load found in people with the pathology. Levels of TDP-43 also tracked with LATE in microglial exosomes, although the differences were not as stark as those in astrocytes.
The scientists repeated the analysis with Cusabio’s commercially available test, which is based on a TDP-43 antibody that recognizes an undisclosed epitope. With this reagent company’s test, only the astrocyte TDP-43 tracked with pathology. Oddly, with the commercial kit, microglial exosomes from people without LATE had more TDP-43 than those with the pathology. Rissman has no explanation for the discrepancy, but said the homemade assay was orders of magnitude more sensitive than the commercial one.
Rissman also reported that the concentration of free TDP-43 in the plasma was similar regardless of LATE, precluding it as a biomarker for the disorder.
Nelson called the potential for a sensitive plasma biomarker for LATE “mind-blowing.” If confirmed, a plasma biomarker for TDP-43 pathology could be useful not only for LATE, but perhaps for FTD as well. TDP-43 pathology underlies about half of FTD cases, while the other half are tauopathies. A TDP-43 marker would be particularly welcome for people with sporadic FTD, who have no mutation to denote their underlying pathology. Since there is no way to tell which neuropathology lurks in their brains, plasma exosome makers could direct them toward tau- or TDP-43-specific clinical trials. Tau-PET tracers do not reliably detect the forms of tau that accumulate in FTD.
Rissman emphasized that his group's findings are preliminary, and must be repeated in larger cohorts. He plans to test plasma from people with other neurodegenerative diseases, including AD and FTD. He also wants to evaluate baseline plasma samples collected in ongoing secondary AD prevention trials, namely A4 and AHEAD 3-45, he told Alzforum. If the assay holds up, researchers will be able to determine whether the TDP-43 biomarker correlated with the trajectory of cognitive decline in placebo groups, and/or the effectiveness of amyloid-targeted therapies in treatment groups after the trials are completed.
Two other potential LATE biomarkers presented at the NIA workshop came from the ranks of structural and functional neuroimaging. Lei Wang of Ohio State University in Columbus shared preliminary data correlating the shape of the hippocampus to the presence of LATE. Using MRI scans taken of participants in the ROS-MAP cohort shortly before they died, Wang found upon autopsy that people who had had LATE tended to have an inward deformation of the hippocampus. In other words, the inner curvature of the seahorse-shaped structure was more concave. This bend correlated with the stage of LATE, and with worse scores on tests of cognitive impairment. Notably, the bend was tied specifically to TDP-43 pathology, occurring independently from hippocampal sclerosis.
Michel Grothe of the Instituto de Biomedicina de Sevilla in Spain described distinctive patterns of hypometabolism in people with LATE versus AD. Recent studies have used FDG-PET to detect metabolic changes in the brain caused by LATE or by hippocampal sclerosis (Botha et al., 2018; Buciuc et al., 2020). To investigate if FDG-PET distinguishes people with LATE and AD, Grothe analyzed scans taken before death among 30 people in the ADNI autopsy cohort, including seven with LATE and no AD pathology, and 23 with plaques and tangles but no LATE. Using these relatively “pure” cases, Grothe found a distinctive, temporolimbic pattern of hypometabolism in LATE, and a more temporoparietal pattern of metabolic dysfunction in AD. Notably, the LATE pattern was independent of hippocampal sclerosis, which was present in three cases.
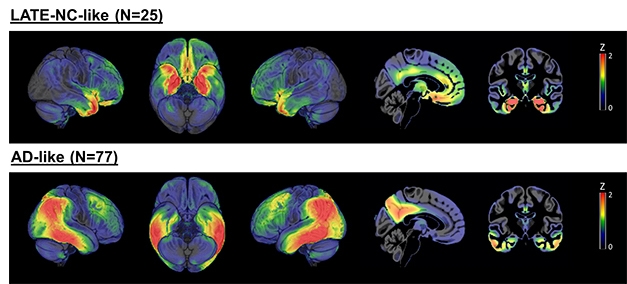
LATE versus AD. A subset of 242 people with a clinical AD dementia diagnosis had either a LATE-like pattern of hypometabolism (top) or AD-like pattern (bottom). Most of the remaining people had patterns that lay somewhere in between. [Courtesy of Michel Grothe, Instituto de Biomedicina de Sevilla, Spain.]
Going back to the whole ADNI cohort, Grothe found that among 242 participants with a clinical diagnosis of AD dementia, 25 had the LATE hypometabolism pattern, hinting that they may have been misdiagnosed, while 77 had the AD-like pattern. The remaining 140 people did not clearly fall into either category. Instead, most fell on a continuum between the AD and LATE patterns, suggesting they may have had a mix of the two pathologies, said Grothe. For the remainder, the metabolic patterns fit nowhere into the proposed continuum.
In keeping with the misdiagnosis idea, Grothe found that people who had more of a LATE pattern had declined more slowly on the MMSE, had closer to normal AD biomarkers, including CSF Aβ42 and p-tau, and were older than those who leaned toward the AD pattern. The proportion of people carrying an ApoE4 allele was smaller among LATE leaners than it was among AD leaners, and they were likelier to carry a TMEM106B risk allele, which has been tied to LATE as well as FTD.
Though he did not present it at the workshop, Grothe told Alzforum that he has examined FDG-PET patterns among people in the ADNI autopsy cohort who had died with a mix of both LATE and AD pathologies. The AD-like hypometabolism pattern predominated, as did plaques and tangles.
Though the findings don’t paint FDG-PET as a clearcut diagnostic tool for LATE, Grothe thinks the scans could prove useful, especially if the LATE to AD continuum can be more clearly defined. While acknowledging there was substantial overlap between people with LATE and AD in these structural and functional imaging measures, Nelson and other researchers were enthusiastic that they might prove valuable upon further refinement.
The meeting agenda, recording, and parallel presentations on LATE are available at LATE 2022.—Jessica Shugart
No Available Further Reading
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.