2016—A Year in Research
Quick Links
Therapeutics
No use soft-pedaling a hard truth: The year ended on a downer. Solanezumab—the therapeutic antibody in line to be the first approved AD drug in well over a decade—failed to significantly slow cognitive decline in people with mild AD and a positive amyloid scan/CSF Aβ profile (see Nov news and Dec conference news). Patients, Lilly, the markets, and the media were predictably disappointed. At the same time, researchers who saw the data at this year’s CTAD conference in December noted positive trends on primary and secondary outcome measures, some of which reached statistical significance by the end of the 18-month Phase 3 trial. Experts in the field concluded that this anti-Aβ antibody had a weak benefit at the dose used. Solanezumab trials in prodromal AD are ongoing, and investigators may up the dose or lengthen those trials to increase the chance of a meaningful slowing of disease.
Meanwhile, sponsors continue to put other immunotherapies through their paces. Biogen published results of the Phase 1b PRIME trial of aducanumab in patients with prodromal or mild AD (see Sep news). PRIME extension data indicated that brain amyloid load continued to fall for another 12 months, and a titration study hinted that ARIA-E can be managed (see Dec conference news).
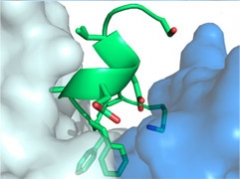
Shape of a Hug.
Solanezumab heavy (blue) and light (white) chains cradle Aβ.
Similarly, a dose-escalation study helped Genentech/Roche choose the sweet spot for Phase 3 trials of their antibody crenezumab. Roche’s anti-Aβ antibody gantenerumab is in Phase 3, and about a dozen additional immunotherapies are in earlier-stage trials. A handful of tau immunotherapies are in Phase 1/2 testing as well.
Leading the pack of non-immunotherapy hopefuls, Merck’s BACE inhibitor verubecestat looked safe in Phase 1 despite concerns about blocking proteolytic cleavage of substrates besides APP (see Nov news). So far, the safety news from the Phase 3 EPOCH trial, which will read out in summer 2017, looks hopeful, with no major adverse events reported (see Oct conference news). Meanwhile, AstraZeneca/Lilly, Biogen/Eisai, Janssen/Shionogi, and Novartis all have BACE inhibitors in the clinic. At a BACE conference, researchers talked about second-generation inhibitors that target BACE1 over BACE2 (see Nov conference news).
Besides solanezumab, other therapeutics that fell on their noses in 2016 in Phase 3 included TauRx’s tau aggregation buster, LMTM, which failed to slow cognitive decline in two AD trials and one FTD trial (see Jul, Sep, and Dec conference news). The end of the road came for idalopirdine, a 5-HT6 serotonin receptor antagonist that did not slow cognitive decline in people with mild to moderate AD (see Sep news). Souvenaid, a drink fortified with antioxidants and omega-3 fatty acids, fell short of showing efficacy in the LipiDiDiet clinical trial (see Mar conference news), though this product is on the market as a food supplement.
In 2016, scientists broadened their attack lines for neurodegenerative disease. One notable here is antisense oligonucleotide (ASO) therapy. Though no ASO’s are being tested in AD yet, nusinersen, an ASO designed to boost transcription of survival motor neuron protein 2, improved motor function in Phase 3 trials of children and infants with spinal muscular atrophy—indeed, the latter trial was halted early because it worked (see Nov news) and was swiftly approved in December. Would that this ever happen in AD! Some clinicians think the findings bode well for preclinical efforts to use ASO to rein in aberrant transcripts and dipeptide repeats encoded by the hexanucleotide repeat expansions in the C9ORF72 gene that cause FTD and ALS (see Apr news).
How about non-drug interventions? Ironically, they could be said to be in a similar spot as some anti-amyloid drugs, in the sense that large trials missed their primary endpoint, yet subgroup analyses brought out signals of a benefit, as in the PreDiva trial (see Aug conference news). Scientists are still refining how best to run trials of multi-domain interventions that bring to bear cardiovascular and metabolic health care and lifestyle advice, to clearly answer if these types of intervention affect Alzheimer’s pathogenesis specifically or if they mitigate broader, age-related risks for dementia.
In 2016, Alzheimer’s therapy research shifted further toward secondary prevention. At full enrollment, DIAN-TU’s first trial of solanezumab and gantenerumab motors on with nary a dropout. DIAN-TU announced this month that Janssen’s BACE inhibitor JNJ-54861911 would be the next drug to be evaluated in this worldwide network of families with autosomal-dominant AD mutations. This drug is also being evaluated in the EARLY (A5) trial of some 2,000 people who are asymptomatic but test positive for brain amyloid (see Aug conference news). As for the ongoing A4 trial of solanezumab, by end of 2016 it had randomized some 800 people at 67 sites and incorporated tau PET into its assessments. The Alzheimer’s Prevention Initiative is still enrolling for its crenezumab trial in Colombian families carrying the presenilin 1 Paisa mutation. For this study, the dose was increased following earlier crenezumab trial results. In 2016, the API also began recruiting for the Generation trial, which aims to enroll 1,300 asymptomatic people carrying two copies of the ApoE4 risk allele, and stave off Alzheimer’s in them with either Novartis’ BACE inhibitor CNP520 or that company’s Aβ vaccine CAD106.
The Generation trial selects candidates by genotyping, an emerging strategy for other neurodegenerative diseases. The GeneMatch program at the Banner Alzheimer’s Institute in Phoenix recruits from among more than 250,000 people who have signed up for the Alzheimer’s Prevention Registry. It uses cheek swabs to identify ApoE4 carriers (see Dec 2015 news). As with stratification based on biomarkers, this strategy comes with the ethical dilemma of disclosure—whether and how to tell people they are heading toward AD. GeneMatch has established a two-stage protocol, first inviting people of all genotypes to an initial screening and then studying the effects of disclosure on those who agree to take part in the trial (ApoE4 carriers and non-carriers alike). Other secondary prevention trials, such as A4, are adopting similar protocols to disclose amyloid status (Sep conference news).
Clinicians studying frontotemporal dementia are watching GeneMatch closely, to see if the strategy might prove useful for the FTD Disorders Registry launched last May. Based in part on the Alzheimer’s Prevention Registry, the FTD registry will provide a resource for FTD clinical trials (see Apr news).

The Global Alzheimer’s Platform
Perhaps the most consequential shift in the AD trials landscape of 2016 was how the field at large is coalescing around internationally coordinated efforts to build standing cohorts of trial-ready study participants for secondary prevention. These people will be characterized for some time before being asked to join trials run by networks of sites that use standardized procedures to make both enrollment and conduct of trials more efficient. As the current Alzheimer’s Disease Coordinated Study is winding down its grant, a Global Alzheimer’s Platform is ramping up (see Aug conference news series).
Biomarkers
The field’s push toward better trials depends critically on appropriate markers. One research goal in 2016 was to fill in the evolving staging diagram of AD with new markers to capture additional aspects of this complex disease. One candidate marker of progression emerged in the form of neurofilament light chain (NfL). Researchers solidified the idea that levels of this axonal protein rise in the blood and CSF of people with AD. NfL also rises in people with other tauopathies and synucleinopathies (see Jun news), but it can be conveniently measured in blood by single molecule immunoassay technology (Simoa), which boasts 25-fold improved sensitivity over traditional ELISAs (see Apr conference news). Simoa will be used to test samples in the DIAN study of early onset AD, and researchers have begun incorporating NfL analysis into clinical trials. The same Simoa test can also be used to measure NfL in mouse models of neurodegenerative diseases, helping bridge the translational gap between animal models and clinical trials. Assays for other markers often don’t work in both human and nonhuman samples.

Axons Tell the Tale.
The rise in neurofilament light reflects axonal damage and swellings (arrowheads), which occur in many brain diseases. [Courtesy of Neuron, Bacioglu et al., 2016.]
The synaptic protein neurogranin burst on the scene in 2015, but this past year, studies of independent data sets strengthened its usefulness for diagnosis and prognosis (e.g., Tarawneh et al., 2016). Neurogranin appears to be more specific to AD than NfL (Wellington et al., 2016).
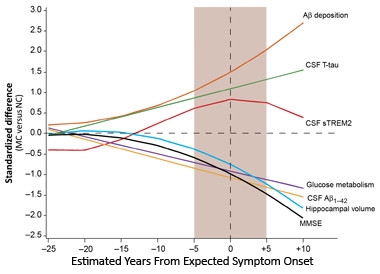
In 2016, eyeballs were trained on the soluble, shed domain of the microglial cell surface receptor, TREM2. Two groups reported higher sTREM2 levels in the CSF of people with mild AD than in control CSF (see Jan news) and a third group saw a rise in CSF sTREM2 in DIAN participants beginning five years before expected onset of symptoms. That’s about when CSF tau begins to rise (see Apr conference news and Dec news). Some five years after symptoms start, soluble CSF TREM2 levels fall again, perhaps reflecting a downturn in microglial activation (see Mar news). CSF levels of another new biomarker, the neuronal calcium sensor VILIP-1, appear to track with sTREM2/tau, rising in the prodromal and later falling in the symptomatic phase of the disease (see Aug conference news).
TREM2 in the Middle.
sTREM2 in spinal fluid peaks around the time CSF tau levels are climbing and memory deficits occur. [Courtesy of Science Translational Medicine/AAAS.]
Developing a plasma test for AD has long been a goal as attractive as it has been elusive. In 2016, researchers identified growth differentiation factor (GDF3) as a potential marker by way of a proteomic screen. Highly expressed in the hippocampus, GDF3 seems to fall in brain and plasma in AD patients (see May news).
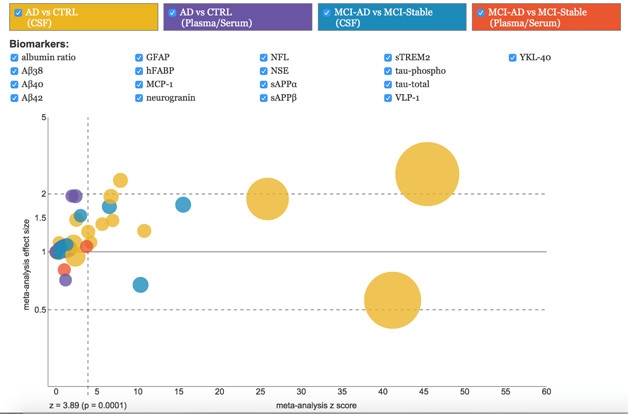
In April, researchers published the methodology underlying AlzBiomarker (see Olsson et al., 2016), the most comprehensive meta-analysis of published Alzheimer’s disease fluid biomarker data to date (see Apr webinar).
In trying to understand which underlying processes previously established CSF markers track, data from the decade-plus BIOCARD longitudinal study bolstered the idea that an uptick in CSF tau heralds cognitive decline in Alzheimer’s (see Apr news). Tau PET helped researchers pinpoint where in the brain tangles correlate strongest with declining neuropsychological performance (see May news). They confirmed that tau PET tracks the progressive spread of tau in the AD brain as established by Braak and Braak (see Mar news). Multimodal imaging correlated brain atrophy with deposition of Aβ and tau in specific regions, suggesting those sites may be where neurodegeneration begins (see Jul news), though how Aβ and/or tau drive neurodegeneration there still remains a nagging question (see Protein Pathology below).

Braak Stages Come to Life. Researchers can now classify people by Braak stages according to in vivo measurements of tau accumulation. [Courtesy of Neuron, Schöll et al.]
UCB-J emerged as a potential imaging biomarker in 2016. A derivative of the anti-epileptic drug levetiracetam, UCB-J binds synaptic vesicle glycoprotein 2A, and could reflect synaptic density, something for which researchers have long wished (see Jul news). Researchers also found a PET tracer for histone deacetylases 1, 2, and 3, which reportedly regulate learning and memory. If this tracer has legs, it might illustrate epigenetic changes in the brain during disease progression (see Aug news).
Protein Pathology
Proteomics revealed that expression of genes that promote protein aggregation ticks up in Braak regions, while expression of genes that prevent aggregates from forming trends down (see Aug news). The finding supports the idea that specific tissue vulnerabilities explain how plaques and tangles spread through the brain, an issue that was hotly debated earlier this year (see Apr webinar). On the other hand, new evidence emerged that neural activity fuels tau spread from cell to cell (see Jun news) and that injecting a trace amount of tau fibrils seeds the propagation of tau aggregates in an otherwise normal brain (see Sep conference news). Specific tau strains favored distinct regions of the brain, buttressing the idea that propagation is an inherent property of certain misfolded proteins, and hinting that different strains of the protein may explain different tauopathies (see Nov news). Researchers debated if Homo sapiens alone is susceptible to Alzheimer’s disease, possibly because other animals resist formation of neurofibrillary tangles (see Dec webinar). A twist on the propagation concept came from a paper claiming that bacterial amyloids kick-start α-synuclein aggregation in the gut, which could spread to the brain (see Oct news).

Taking up Tau. In microfluidic chambers, tau (red) migrates along axons to adjacent cambers to be taken up by tau knockout neurons (green). [Courtesy of Wu et al., Nature Neuroscience.]
In 2016, researchers identified new modes of tau regulation. Nuak1, a homologue of AMP-activated protein kinase, reportedly phosphorylates the microtubule binding protein, preventing its degradation and causing its accumulation (see Oct news). Reducing Nuak1 rescued a mouse tauopathy model. Others challenged the notion that tau phosphorylation is always bad, claiming that p38γ prevents Aβ-mediated excitotoxicity by phosphorylating tau at threonine 205 (see Nov news). If these twists and turns of tau regulation aren’t hard enough to follow, try this: Tau harboring the alanine-152-threonine mutation wreaks havoc without aggregating, possibly via soluble oligomers. Mice with this variant developed scant tangles but rampant neurotoxicity, degeneration, and inflammation (see Mar news).

Snagged.
Scanning electron microscopy revealed fibrous material (red arrows) around yeast incubated with H4 cells that produce Aβ (right). [Courtesy of Science Translational Medicine/AAAS.]
A year without new insights into Aβ dynamics would be rare indeed. In 2016, praise rolled in for a paper describing how a sorting signal in presenilin-2 directs this protease to lysosomes, where it generates a pool of intracellular Aβ enriched with Aβ42. This underscored the importance of the endolysosomal system in AD and renewed interest in intraneuronal Aβ (see Jun news). Researchers fingered mutant presenilin for aberrant proteolytic cleavage of the endoplasmic reticulum calcium sensor STIM-1, disrupting Ca2+ signaling and shrinking dendritic spines (see Sep news). Two papers documented that supposedly inactive presenilin-1 mutants actually generate the potently toxic Aβ43, further contradicting the loss-of-function presenilin function hypothesis (see Apr news). As for Aβ itself, the idea that it fights microbes returned with new data in worm and mouse models of AD (see May news), though it has not caught on broadly among many labs yet.
Liquid Proteins?
The concept of liquid-phase separation of proteins took the field by storm in 2015 (see Oct 2015 webinar) and gelled further in 2016. Building on the idea that certain proteins condense to form liquid droplets in cells, scientists reported that poly-dipeptide repeats translated from the hexanucleotide expansion in the C9ORF72 gene disrupt these membrane-less organelles (see Oct news). Specifically, the dipeptide repeats bind to proteins with low-complexity domains, which tend to morph into liquid droplets. They include TDP43, FUS, hnRNPA1, and other proteins linked to neurodegeneration. Scientists believe that disrupting the liquid organelles interferes with normal cellular processes, explaining at least in part how the hexanucleotide repeats cause ALS and FTD. Yet poly glycine-alanine, one of five potential dipeptides the repeats encode, seems insufficient to explain ALS because it does not cause the accumulation of TDP43, a hallmark of the disease (see Mar news). Along the same lines, ALS-linked mutations in the low-complexity domain of TDP43 also suppressed liquid phase separation, allowing the protein to form toxic aggregates instead (see Sep news).
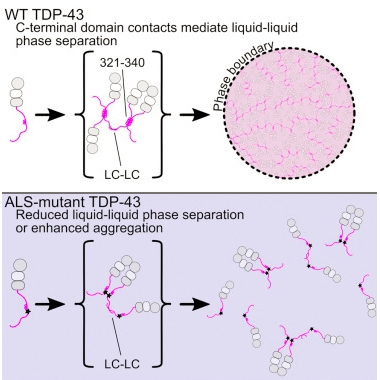
Protein Phase.
TDP-43’s C-terminal helices (pink) form an orderly assembly (top) that allows liquid-liquid phase separation. Mutations (black stars) disrupt this process, promoting protein aggregation instead (bottom). [Courtesy of Structure, Conicella et al.]
Researchers continue to debate whether sense and antisense RNAs transcribed from the C9ORF72 hexanucleotide expansion form aggregates, and to weigh evidence for a toxic role of these RNAs in neurodegeneration. RNAs containing these expansions, or expansions of the trinucleotide CAG repeat found in the huntingtin gene, were spotted traveling to neuronal extremities, where they interfered with local protein translation, causing dendritic arbors to wither (see Feb news). Quelling the transcription elongation factor Spt4, which helps the ribosome traverse repetitive sections of DNA, suppressed sense and antisense C9ORF72 transcripts at the source (see Aug news).
In other C9ORF72 news, a normal function for the protein is coming into view. It appears to modify cofilin to regulate actin dynamics in motor neurons. Knocking down the native protein shrank mouse motor neuron axons, suggesting that the hexanucleotide expansion in the gene may cause a toxic loss of function (see Oct news). The finding jibes with prior work suggesting C9ORF72 belongs to a protein family whose members modify guanine nucleotide exchange factors, aka GEFs, which regulate cofilin kinases (see Jan 2013 news).
Glia
Interest in microglia, the brain’s innate immune cells, exploded in 2016 (see Jun conference series). Scientists are digging into how microglia interact with Aβ, and what role, if any, TREM2 plays in that. In a finding germane to passive immunotherapy trials, antibodies generated by the adaptive immune system stimulated mouse microglia to phagocytose and engulf Aβ (see Feb news). Without TREM2, microglia poorly phagocytosed Aβ-antibody complexes, perhaps explaining why mutations in this cell surface receptor increase a person’s risk for AD (Jul news). While these studies support the theory that TREM2 activation promotes microglial uptake of Aβ, evidence for an alternative hypothesis emerged when super resolution microscopy revealed microglia encapsulating plaques, limiting diffusion of Aβ (see May news).
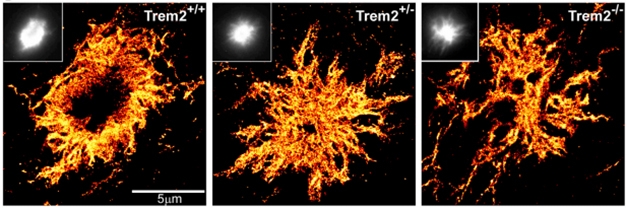
Plaque Anatomy. STORM shows that with TREM2 in short supply, plaques contain more diffuse Aβ. [Courtesy of Jaime Grutzendler and Neuron.]
In other glia news, the idea that microglia take an active role in synaptic pruning, and that this may be important in early AD, gained further traction (see Apr news). A method for purification of mature human astrocytes led to a transcriptional database that will help studies of these abundant though poorly understood cells (see Jan news).
Disease Models
Many labs have tried to develop mouse models of ALS/FTD based on loss or gain of function of the C9ORF72 gene (see Oct 2015 conference news), and in 2016, three genetic knockout models came out. All three had immune problems, growing huge spleens and enlarged lymph nodes. They had intense inflammatory responses but no obvious neural problems (see Mar news). A fourth knockout had even worse immune problems, dying young from aggressive auto-immunity (see Jul news). The findings jibe with reports of conditional knockouts that seem to get by without C9ORF72 in the nervous system (see Jun 2015 news). In contrast, mice expressing a bacterial artificial chromosome carrying human C9ORF72 with various length hexanucleotide repeats do recapitulate facets of ALS/FTD, including dipeptide repeat aggregates, TDP-43 inclusions, denervated neuromuscular junctions, movement problems, and signs of anxiety (see May news).
A different type of ALS model debuted in November. Mice expressing a mutated version of the human ubiquilin 2 gene accumulated TDP43 in the spinal cord, leading to motor neuron degeneration (see Nov news). Mutations in UBQLN2 have been linked to familial ALS/FTD. In March, Alzforum released an interactive guide to 27 different ALS mouse models (see Research Models).
In the AD field, the pros and cons of overexpression versus knock-in models continue to be actively debated. The former produce some artifacts related to overexpression or gene disruption, whereas APP knock-ins have a mild phenotype that makes them unsuitable for quick screening experiments (see Dec conference news). Nevertheless, hundreds of laboratories around the globe are now raising APP knock-in mice developed in Japan, and initial publications are beginning to question prior conclusions based on overexpression models (see Sep news). Next up, scientists are making tau knock-in models, and preliminary results suggest that crossing the tau knock-ins with the APP knock-ins results in mice that more faithfully recapitulate AD pathology, featuring hyperphosphorylated, insoluble tau, and neuronal death (see Dec conference news).
Genetics
Now that large genome-wide association studies have all but exhausted the search for common AD risk variants, researchers use new methods to understand those variants and to uncover new ones. Recent data support the idea that different polymorphisms influence amyloidosis, neurofibrillary tangles, and brain atrophy. One research group took a pleiotropic approach. Using data from the International Genomics of Alzheimer’s Project (IGAP), they identified eight variants linked to both autoimmunity and AD. Two increased risk for dementia; six seemed protective. The former correlated with tangle pathology but not amyloidosis (see Apr news). Others went straight for biomarkers, using IGAP data to calculate a polygenic risk score (PGRS), and testing how it tracked with both amyloid and metabolism. The upshot: The score predicted only a decline in baseline metabolism, particularly in certain brain regions (see Sep conference news).
Data from ADNI suggested that the effect of genetic risk variants may depend on disease stage. Some of the top 20 GWAS hits correlated with atrophy only in prodromal or full-blown AD, but not both. ADNI data also led to new variants that correlated with cortical thinning (see Sep conference news). In ADNI, PGRS associated with abnormally low hippocampal volume and poorer performance on memory tests among otherwise healthy old adults, and it predicted decline of memory and executive function (see Jul news).
The search for rare polymorphisms with stronger effects than the common variants identified in GWAS got a boost from the Exome Aggregation Consortium (ExAC), which announced that it had assembled full exome sequences from 60,706 people worldwide (see Aug news).
Funding/Policy
The outlook for research brightened considerably in 2016. The U.S. Senate earmarked $1.4 billion for dementia research in 2017, an increase of 40 percent (see Jun news). This followed a 60 percent hike in funding in 2015. Up from the dog days of 2010-11 when it was 3 percent, the NIA’s current pay lines reflected the boost, funding about 22 percent of proposals smaller than $500,000 (26 percent for new or early stage investigators), and 19 percent of larger grants (23 percent for those younger investigators). However, the Brexit vote troubled researchers on both sides of the Atlantic. Will E.U. funding for research institutions in Britain dry up? And what about international collaboration (see Jul news)? Prime Minister Theresa May promised to outline proposals for exit negotiations in early 2017, and to trigger negotiations with the EU by March. In the meantime, the British Medical Research Council chose a Belgian, Bart De Strooper, to head its Dementia Research Institute (see Dec interview). Announced in May, the £250 million DRI was the brainchild of former Prime Minister David Cameron, who campaigned against Brexit (see May news).
Dementia on the Decline?
A steady decline in dementia incidence—initially captured in Sweden, England, and Germany—seems afoot in the United States, as well. Three decades of data from the Framingham Heart Study saw incidence fall by nearly half since the late 1970s (see Feb news), and data from the Health and Retirement Study backed this up with a 24 percent drop in prevalence between 2000 and 2012 (see Nov news). The HRS samples data from people of all socioeconomic backgrounds across the United States. Researchers are unsure why the rates are falling so steeply. Better cardiovascular health, more education, and healthier all-around lifestyle may partly explain this, but whatever the reason, they now predict a million fewer cases in the United States in the next two decades than they had before. That said, as baby boomers age, the United States still faces 14 million AD cases by 2050, and incidence rates are expected to climb in low- and middle-income countries as their populations age.
Microbiome and the Brain
Call it a gut feeling, but do our multitudinous intestinal flora have more to say about brain health than we realized? An earlier paper outlined how short-chain fatty acids released by intestinal bacteria bolster microglia in the brain (see Jun 2015 news), and this past month came news of a nefarious twist—those microbial fatty acids might stoke the flames of Parkinson’s by increasing brain inflammation and promoting aggregation of α-synuclein. Intriguingly, the microbiota in PD patients seem distinct from those of healthy controls, the former exacerbating PD-like pathology when inoculated into mouse models (see Dec news). This news came on the heels of word that an E. coli amyloid called curli cross-seeded aggregation of α-synuclein in the guts of aged rats. Synuclein in the gut then travelled up the vagus nerve to the brain, where it seeded further aggregation, the authors proposed (see Oct news). The findings tie the microbiome to the hypothesis that Parkinson’s starts in neurons of the intestine and spreads from there to the brain.

Microbes Matter.
Typical microbiota (left panel) secrete enough SCFAs to activate microglia that can exacerbate PD, while a lack of microbes (middle panel) prevents this process. Something about microbes derived from PD patients (right panel) ramps up disease progression even more than typical microbiota. [Courtesy of Sampson et al., Cell 2016.]
There is not much evidence that microbes influence Aβ aggregation, though feeding antibiotics to male APP/PSEN1 mice halved plaque burden in their brains, and infecting mouse brains with Salmonella seeded plaques (see May conference news). The latter observation fits with the idea that Aβ evolved as an antimicrobial (see Protein Pathology, above).
Pathology/Staging
Diagnostic and staging criteria for AD have been transformed in the last decade, and researchers are fine-tuning them as new biomarker data come in. Researchers proposed a revision of the NIA/AA diagnostic criteria published in 2011, when biomarkers data were first—and tentatively—woven into the guidelines. The current revision proposes that biomarkers should be the sole diagnostic arbiters, at least for research purposes. This latest iteration of the research criteria would rely on a classification scheme dubbed A/T/N. It assigns positive or negative scores to amyloid pathology, tau pathology, and neurodegeneration, respectively (see Aug news). The A/T/N scheme separates tau from neurodegeneration—a major departure from the past. This may help clinicians better define pathology, especially in people with SNAP, aka suspected non-Alzheimer pathophysiology, who show signs of neurodegeneration but test negative for Aβ and do not progress like AD patients do.
It remains unclear how this A/T/N classification would alter current diagnosis of AD. Researchers in DIAN are testing the scheme by combining more traditional markers with flortaucipir PET (see Aug news). Some researchers dislike that it requires binary yes/no demarcations for parameters that range widely, especially since thresholds for those measures are arbitrary.
Vascular Disease and Dementia
In recent years, connections have strengthened between vascular risk factors and dementia. One controversial paper extended the idea, claiming that in late-onset AD, vascular pathology emerges first, even before Aβ pathology, (see Jul news). Others questioned the methodology, noting that the data contradict trajectories documented in countless other studies. Researchers agreed that vascular pathology may predispose people to AD, and in 2016 reported that cerebral microbleeds drive up risk for dementia (see Jun news), that blood platelets may seed aggregation of Aβ (see Jun news), and that dementia and vascular pathology might form a vicious circle, since a buildup of tau in the mouse brain appears to restrict blood flow (see May conference news).
Scientists are beginning to get a handle on the vasculature in dementia by focusing on specialized pericytes, astrocytes, and teeny blood vessels of the neurovascular unit. Meanwhile, molecules floating around in blood cannot be ignored. According to one mouse study, ApoE in plasma, but not brain, restores synaptic deficits and improves learning and memory, suggesting that peripheral ApoE may prevent breakdown of the blood-brain barrier or regulated lipids essential to brain function (see Oct news). Along the same vein, more evidence surfaced that molecules circulating in the plasma of young mice rejuvenated old brains. In parabiosis, where the circulatory systems of two mice are temporarily spliced together, young wild-type mice restored synaptophysin levels in older APP transgenic mice with rampant amyloid plaques. The plaques stayed put, suggesting that something in young mouse blood directly soothes synapses. What that is remains a mystery (see Sep news).
Sleep, Network Activity, and Dementia
Before you nod off, cast a drowsy eye over links between neurodegenerative diseases and modulation of network activities, including sleep. Though it appears essential for memory consolidation and clearance of Aβ, sleep and its regulation remain poorly understood. Surprisingly, changes in ion levels in the brain interstitial fluid may control the sleep/wake cycle. Those fluctuations can also change the osmotic pressure in the brain, modulating glymphatic clearance of solutes, including Aβ, researchers proposed. Others tied sleep deprivation to clogged Virchow-Robin spaces, those gaps between blood vessels and brain cells that help drain interstitial fluid, and to the protein synthesis that is essential for memory consolidation (see May news).
Specific network activities may be essential for memory consolidation. Low-frequency theta rhythms of 4 to 7 Hz cement memories during REM sleep (see May news), while sleep in general appears to strengthen synaptic connectivity (see Kuhn et al., 2016). Perhaps most intriguing, though not directly sleep-related, was a year-end report that strengthening gamma oscillations in mouse models of AD suppressed Aβ production in the brain and helped clear plaques, possibly by activating microglia (see Dec news). In people, gamma rhythms weaken with AD.—Tom Fagan and Gabrielle Strobel
References
Therapeutics Citations
- Solanezumab
- Aduhelm
- Crenezumab
- Gantenerumab
- Verubecestat
- Idalopirdine
- Souvenaid
- Atabecestat
- Umibecestat
- Amilomotide
- Levetiracetam
News Citations
- Lilliputian Effect Size Fells Phase 3 Trial of Solanezumab, Leaving Its Future Uncertain
- CTAD: Solanezumab Seen to Nudge AD Ever so Slightly
- Paper Alert: Aducanumab Phase 1b Study Published
- Much ‘Adu’ About a Little: Phase 1 Data Feeds the Buzz at CTAD
- Paper Alert: Verubecestat Preclinical and Phase 1 Data Published
- At 2nd Kloster Seeon Meeting, Renewed Optimism for Targeting BACE1
- Will Next-Gen BACE Inhibitors Dodge Side Effects?
- In First Phase 3 Trial, the Tau Drug LMTM Did Not Work. Period.
- First Round of FTD Therapeutics Fell Short, But Many More Are Up and Running
- STARSHINE Trial Loses its Luster
- Souvenaid Trial Missed Primary, Partially Met Secondary Endpoints
- Positive Trials of Spinal Muscular Atrophy Bode Well for Antisense Approach
- Paper Alert: Could Antisense Therapy One Day Squelch Toxic Repeats in ALS or FTD?
- PreDIVA Trial Falls Short
- New Ways to Target Aβ and BACE Show Promising Phase 1 Data
- GeneMatch Registry Recruits Subjects for Prevention Trials
- To Know or Not to Know: Trial Participants Confront the Question
- Drug Trials in Frontotemporal Dementia: Can Field Push Forward Together?
- Coming to a Center Near You: GAP and EPAD to Revamp Alzheimer’s Trials
- Blood NfL Looks Good as Progression and Outcome Marker
- WANTED: Biomarkers for Drug Trials in Frontotemporal Dementia
- TREM2 Goes Up in Spinal Fluid in Early Alzheimer’s
- DIAN Longitudinal Data Say Cognition Goes Earlier Than Previously Thought
- Paper Alert: Slotting TREM2 into Alzheimer’s Pathogenesis
- Microglial Marker TREM2 Rises in Early Alzheimer’s and on Western Diet
- Plasma Proteomics Study Hints at New Player in Alzheimer’s
- CSF Tau Rivals Aβ for Predicting Cognitive Decline
- On Multiple Marker Analysis, Tangles Track Best With Functional Decline
- Tau PET Aligns Spread of Pathology with Alzheimer’s Staging
- Do Temporal Lobe Tangles and Cortical Plaques Together Bring on Alzheimer’s?
- Next Up for Human Brain Imaging: Synaptic Density?
- Epigenetic Tracer Uncovers Patterns of Healthy Gene Regulation
- Aggregation-Prone Gene Expression Signature Mapped in Brain
- Excited Neurons Release More Aberrant Tau
- New Data Reinforces Concept of Protein Propagation
- More Evidence That Distinct Tau Strains May Cause Different Tauopathies
- Could Bacterial Amyloid Trigger Parkinson’s Pathology?
- New Target for Tauopathies? Blocking Nuak1 Could Reduce Tau Build-Up
- Is Tau Phosphorylation All Bad?
- A152T: A Different Kind of Tau Mutation
- Lodged in Late Endosomes, Presenilin 2 Churns Out Intraneuronal Aβ
- Mutant Presenilin Skews Calcium Homeostasis by Chomping on ER Sensor
- Pathogenic Presenilin Mutations Generate Aβ43
- Like a Tiny Spider-Man, Aβ May Fight Infection by Cocooning Microbes
- ALS Research ‘Gels’ as Studies Tie Disparate Genetic Factors Together
- C9ORF72 Dipeptide Repeat Not Enough for Motor Neuron Disease
- Helical Tail Holds Sway Over TDP-43 Packaging
- Repeat RNAs Hitchhike to the Ends of Neurons, Attacking Neurites
- Tackling Mutant C9ORF72 Transcripts at the Source
- Grow that Axon: C9ORF72 Function Revealed?
- C9ORF72 Function: Is the ALS Protein a Membrane Traffic Cop?
- Bon Appétit: Endogenous Antibodies Prod Microglia to Eat Aβ Deposits
- TREM2 Helps Phagocytes Gobble Up Aβ Coated in Antibodies
- Barrier Function: TREM2 Helps Microglia to Compact Amyloid Plaques
- Paper Alert: Microglia Mediate Synaptic Loss in Early Alzheimer’s Disease
- Purification of Adult Human Astrocytes Shows: They Are Unique
- C9ORF72 Mice Point to Gain of Toxic Function in ALS, FTD
- C9ORF72 Knockout Causes Inflammation, not Neurodegeneration
- Paper Alert: Autoimmunity in Another C9ORF72-Deficient Mouse Strain
- No C9ORF72, No Problem: Knockout Mouse Neurologically OK
- New C9ORF72 Mice Develop Symptoms Resembling ALS/FTD
- ALS-FTD Mouse Model Develops Motor Neuron Disease
- Knock-In Alzheimer’s Mice Catch on More Broadly in the Field
- Do APP Knock-ins Call Overexpression Models of AD into Question?
- Next-Generation Mouse Models: Tau Knock-ins and Human Chimeras
- New Genetic Method Connects Immune Genes to Alzheimer’s
- Amyloid and Neurodegeneration Have Different Underlying Genetics
- Are Early Harbingers of Alzheimer’s Scattered Across the Genome?
- Flood of Exomes Brings Genetic Variation into Focus
- Possible Boost for U.S. Alzheimer’s Research on Horizon
- Brexit Could Threaten Neurodegenerative Disease Research in Europe
- Post-Brexit U.K. Picks Belgian to Run Its Flagship Dementia Research Institute
- New Dementia Research Institute Announced for the U.K.
- Falling Dementia Rates in U.S. and Europe Sharpen Focus on Lifestyle
- U.S. Dementia Rates Fall
- To Be Hale and Hearty, Brain Microglia Need a Healthy Gut
- Do Microbes in the Gut Trigger Parkinson’s Disease?
- Microbial Hypotheses Intrigue at Zilkha Alzheimer’s Meeting
- Staging of Alzheimer’s, the Second: Neurodegeneration Does Not Equal Tauopathy
- Brain Imaging in DIAN: Atrophy Rates—Check. Tau PET—Not Yet.
- LOAD of Data Place Vascular Malfunction as Earliest Event in Alzheimer’s
- Microbleeds in the Brain Portend Dementia
- Do Platelets Provide a Platform for Aβ Aggregation?
- What’s Up With the Vasculature in Dementia?
- Distinct Roles for Brain and Blood ApoE in Neuron Health?
- Young Blood a Boon for APP Mice
- Sleep and Brain Cleansing—Fresh Insights into Regulation and Disruption
- In mTORC and Theta Rhythms, New Clues to How Sleep Locks Down Memories
- Flashy Treatment Synchronizes Neurons, Lowers Aβ in Mice
Biomarker Meta Analysis Citations
Webinar Citations
- Learn About AD Biomarker Meta-Analysis on AlzBiomarker Database
- Webinar: Pathogenic Protein Spread? Let’s Think Again
- Is Alzheimer’s Disease a Uniquely Human Disorder?
- Fluid Business: Could “Liquid” Protein Herald Neurodegeneration?
Mutations Citations
Conference Coverage Series Citations
Research Models Citations
Paper Citations
- Tarawneh R, D'Angelo G, Crimmins D, Herries E, Griest T, Fagan AM, Zipfel GJ, Ladenson JH, Morris JC, Holtzman DM. Diagnostic and Prognostic Utility of the Synaptic Marker Neurogranin in Alzheimer Disease. JAMA Neurol. 2016 May 1;73(5):561-71. PubMed.
- Wellington H, Paterson RW, Portelius E, Törnqvist U, Magdalinou N, Fox NC, Blennow K, Schott JM, Zetterberg H. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology. 2016 Mar 1;86(9):829-35. Epub 2016 Jan 29 PubMed.
- Olsson B, Lautner R, Andreasson U, Öhrfelt A, Portelius E, Bjerke M, Hölttä M, Rosén C, Olsson C, Strobel G, Wu E, Dakin K, Petzold M, Blennow K, Zetterberg H. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. Lancet Neurol. 2016 Jun;15(7):673-84. Epub 2016 Apr 8 PubMed.
- Kuhn M, Wolf E, Maier JG, Mainberger F, Feige B, Schmid H, Bürklin J, Maywald S, Mall V, Jung NH, Reis J, Spiegelhalder K, Klöppel S, Sterr A, Eckert A, Riemann D, Normann C, Nissen C. Sleep recalibrates homeostatic and associative synaptic plasticity in the human cortex. Nat Commun. 2016 Aug 23;7:12455. PubMed.
Other Citations
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.