Lack of C9ORF72 Protein Renders Neurons More Vulnerable to Degeneration
Quick Links
Ever since repeat expansions in the C9ORF72 gene were identified as a major cause of amyotrophic lateral sclerosis and frontotemporal dementia, researchers have been debating whether the disease is caused by loss of the normal protein or accumulation of toxic aggregates. Most previous data has trended toward the latter. However, a new study in the February 5 Nature Medicine makes the case that both mechanisms contribute, with a drop in levels of C9ORF72 protein leaving neurons more susceptible to the toxic effects of the repeat expansion.
- Loss of C9ORF72 protein renders motor neurons susceptible to excitotoxicity.
- It also interferes with lysosomal maturation, allowing toxic DPRs to build up.
- Thus, loss- and gain-of-function mechanisms may together bring on ALS.
Researchers led by Justin Ichida at the University of Southern California in Los Angeles used pluripotent stem cells to show that induced motor neurons with little C9ORF72 died more readily than control cells when exposed to stressors such as nutrient depletion or excess glutamate. Adding back a normal C9ORF72 gene rescued survival. In addition, low C9ORF72 levels hampered endosomal trafficking, leading to a dearth of lysosomes. That in turn allowed toxic dipeptide repeat proteins (DPRs) made from the expanded RNA to pile up. Inhibiting an endosomal kinase, PIKFYVE, restored lysosomal maturation, reduced excitotoxicity, and lowered the number of aggregates. “PIKFYVE inhibition appears to modulate several ALS disease processes, and may have therapeutic potential,” Ichida told Alzforum.
Other researchers were enthusiastic. “This is a landmark study,” Jiou Wang at Johns Hopkins University in Baltimore wrote to Alzforum. He praised the thoroughness of the experiments and controls, noting that they represent a heroic effort to address multiple aspects of C9ORF72-associated toxicity. Robert Baloh at Cedars-Sinai Regenerative Medicine Institute, Los Angeles, agreed. “[This] adds further evidence that the disease cannot be simply looked at from the perspective of one potential toxic mechanism, such as RNA granules or dipeptide repeats, but rather should be looked at as resulting from a combination of gain- and loss-of-function effects within and across cell types in the nervous system,” he wrote (see comment below).
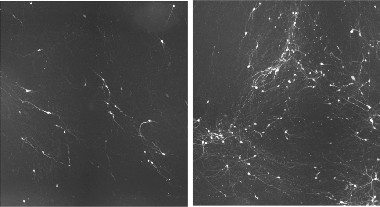
Motor Neuron Rescue.
Induced neurons made from patients with the C9ORF72 expansion (left) die off after one week in culture, but treatment with a PIKFYVE inhibitor preserves them (right). [Courtesy of Shi et al., Nature Medicine.]
Gain-of-function mechanisms have garnered the most attention from researchers, with numerous studies linking C9ORF72 expanded RNA foci and DPRs to toxicity in cell cultures (Dec 2014 news; Jul 2017 news; Feb 2018 news). However, C9ORF72 expansions have little effect in many mouse models, suggesting other factors may contribute to the disease, as well (May 2016 news). Meanwhile, the repeat expansion squelches production of C9ORF72 protein, raising the possibility that loss of normal protein function could factor into the disease. Researchers have paid less attention to this idea, because C9ORF72 knockout mice do not lose motor neurons or have movement problems (Jun 2015 news).
Stress Unmasks C9ORF72 Haploinsufficiency
Ichida and colleagues decided to take a closer look at C9ORF72’s role. Joint first authors Yingxiao Shi and Shaoyu Lin confirmed that induced motor neurons (iMNs) generated from ALS and FTD patients carrying the expansion made about half as much C9 protein as did iMNs from controls. Under normal culture conditions, these C9 patient iMNs survived as well as control iMNs, the authors found. Under stress from nutrient withdrawal or excess glutamate, however, C9 cells died off more quickly.
Was lack of C9ORF72 protein to blame for this vulnerability? To test this, the authors upped C9ORF72 expression in patient and control iMNs by expressing the normal gene off a retrovirus. This rescued survival in patient iMNs and had no effect on controls. Then, the researchers knocked out one or both copies of C9ORF72 in control iMNs. These cells died as quickly as C9 patient iMNs upon exposure to excess glutamate. In addition, transcriptional profiles from C9-deficient iMNs, C9 patient iMNs, and postmortem tissue from C9 patients all resembled each other, but varied from the profile of healthy controls.
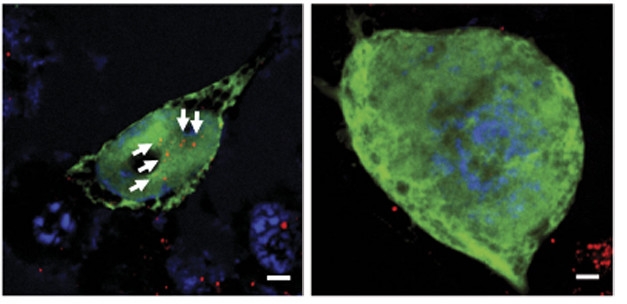
More C9ORF72, Fewer Deposits. Motor neurons with C9 expansions (left) accumulate DPRs (red, white arrows), but adding C9 protein clears these deposits (right). [Courtesy of Shi et al., Nature Medicine.]
Gain and Loss of Function Both Converge on Excitotoxicity
Ichida and colleagues next tried to dissect how low C9ORF72 levels caused problems. Because of the glutamate sensitivity of patient iMNs, they measured NMDA and AMPA glutamate receptors on the cell surface. They found that C9 patient iMNs had about twice as many receptors as controls. Spinal motor neurons from a C9 knockout mouse, as well as motor neurons from patient postmortem tissue, had double the receptors as well. The reason for this glut is unclear, although C9ORF72 has been reported to affect vesicle trafficking, which may influence receptor movement (Farg et al., 2014; Aoki et al., 2017).
The motor neurons’ glutamate sensitivity matters, because ALS patients have high levels of glutamate in cerebrospinal fluid. Some researchers believe this is caused by DPRs that are taken up into astrocytes, where they interfere with splicing of an astrocytic glutamate transporter, rendering the glia unable to mop up extracellular glutamate (Lin et al., 1998; Aug 2014 news; Jun 2017 news). Thus, loss of C9ORF72 may conspire with DPR toxicity to kill off motor neurons, the authors proposed.
In fact, glutamate sensitivity was specific to motor neurons. When the authors differentiated iPSCs from people with C9 expansions into dopaminergic neurons, those cells survived as well as controls in the presence of high glutamate levels. Ichida noted that motor neurons are believed to be more susceptible to excitotoxicity than other neurons due to poor calcium buffering (Van Den Bosch et al., 2000; Van Damme et al., 2007; Bogaert et al., 2010). Motor neurons selectively degenerate in ALS.
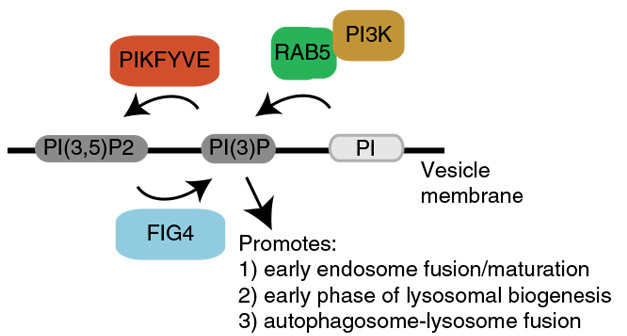
Lysosomal Chokepoint. The kinase PIKFYVE and the phosphatase FIG4 exert opposing effects on endosomal lipids that regulate their maturation into lysosomes. [Courtesy of Shi et al., Nature Medicine.]
Autophagy, Again
Excitotoxicity was not the only problem for C9ORF72-deficient iMNs. These cells also had about a third fewer lysosomes than controls. So did spinal motor neurons from the C9ORF72 knockout mouse. Expressing normal C9 in patient iMNs and C9 knockout iMNs restored lysosome numbers.
A lack of lysosomes might make motor neurons less able to degrade dipeptide repeat protein aggregates, the authors reasoned. In keeping with this idea, C9-deficient and patient iMNs cleared exogenous DPR deposits more slowly than control iMNs did, and the cells died faster. Conversely, adding C9ORF72 to patient iMNs lowered the number of DPR aggregates.
How might C9 control lysosome quantity? Ichida and colleagues found that about 80 percent of vesicle-bound C9ORF72 was located in early endosomes. There, it bound to the early endosomal marker, early endosome antigen 1. EEA1 in turn binds the lipid phosphatidylinositol 3-phosphate (PI3P) to drive endosomal maturation to lysosomes, as well as the fusion of those vesicles with autophagosomes. The findings suggested that a lack of C9 could block lysosomal maturation.
Previous studies have already implicated C9 in regulating autophagy (Webster et al., 2016; Sullivan et al., 2016). Some groups have found that stimulating autophagy boosts survival in ALS models, although others caution that more autophagy is not always better (May 2017 news; Sep 2017 news).
A Therapeutic Angle Emerges
The authors screened 800 small molecules for compounds to improve C9 iMN survival. Among the hits was an inhibitor of PIKFYVE, which localizes to early endosomes. PIKFYVE phosphorylates PI3P, converting it into an inactive form, thereby inhibiting endosomal fusion with lysosomes. Hence, inhibiting PIKFYVE stimulates autophagy. Intriguingly, the phosphatase FIG4, also found in early endosomes, opposes PIKFYVE function by activating PI3P (see image above). Loss-of-function mutations in FIG4 cause ALS, highlighting the importance of this pathway to the disease (Chow et al., 2009).
The PIKFYVE inhibitor rescued C9 patient iMN survival in a dose-dependent manner (see image above). The authors then tested a more potent and structurally distinct PIKFYVE inhibitor, apilimod. Apilimod has demonstrated safety in clinical trials for autoimmune disorders. It likewise rescued survival of C9 iMNs, and lowered their NMDA and AMPA receptor levels. In mouse models, apilimod blocked neurodegeneration and lowered DPR aggregates in hippocampal neurons.
“We are excited about this pathway because of the genetic link to the FIG4 form of ALS, and because PIKFYVE inhibition has the ability to rescue both the loss- and gain-of-function problems,” Ichida told Alzforum. He co-founded a company, AcuraStem in Monrovia, California, to develop PIKFYVE inhibitors for clinical trials. Company researchers have found that the body quickly metabolizes apilimod, perhaps explaining its failure in previous clinical trials. They are working on ways to stabilize the molecule. Baloh is an adviser to AcuraStem.
Shawn Ferguson at Yale University, New Haven, Connecticut, noted that the approved ALS drug riluzole inhibits excitotoxicity, and suggested comparing the effects of PIKFYVE inhibition to riluzole in cell cultures. “Significant follow-up is required to determine whether the beneficial effects observed by Shi et al. in relatively acute assays in very specific models can translate into significant therapeutic benefits in human disease that would exceed those seen with existing glutamate blockers such as riluzole,” Ferguson wrote (see comment below).—Madolyn Bowman Rogers
References
News Citations
- Live-Cell Studies Blame Arginine Peptides for C9ORF72’s Crimes
- Barring Export of Expanded Repeat RNAs Tames Their Toxicity
- Are ALS Dipeptide Repeat Ribbons Entangling Proteasomes?
- New C9ORF72 Mice Develop Symptoms Resembling ALS/FTD
- No C9ORF72, No Problem: Knockout Mouse Neurologically OK
- C9ORF72 Killer Dipeptides Clog the Nucleolus
- When C9ORF72 Silences U2, Spliceosomes Can’t Find What They’re Looking For
- Certain Cancer Drugs Held Out as Prospects for ALS Therapy
- From Protector to Instigator, Autophagy Makes About-Face in ALS
Therapeutics Citations
Paper Citations
- Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson PA, Blair IP, Soo KY, King AE, Atkin JD. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014 Jul 1;23(13):3579-95. Epub 2014 Feb 18 PubMed.
- Aoki Y, Manzano R, Lee Y, Dafinca R, Aoki M, Douglas AG, Varela MA, Sathyaprakash C, Scaber J, Barbagallo P, Vader P, Mäger I, Ezzat K, Turner MR, Ito N, Gasco S, Ohbayashi N, El Andaloussi S, Takeda S, Fukuda M, Talbot K, Wood MJ. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain. 2017 Apr 1;140(4):887-897. PubMed.
- Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, Rothstein JD. Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron. 1998 Mar;20(3):589-602. PubMed.
- Van Den Bosch L, Vandenberghe W, Klaassen H, Van Houtte E, Robberecht W. Ca(2+)-permeable AMPA receptors and selective vulnerability of motor neurons. J Neurol Sci. 2000 Nov 1;180(1-2):29-34. PubMed.
- Van Damme P, Bogaert E, Dewil M, Hersmus N, Kiraly D, Scheveneels W, Bockx I, Braeken D, Verpoorten N, Verhoeven K, Timmerman V, Herijgers P, Callewaert G, Carmeliet P, Van Den Bosch L, Robberecht W. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc Natl Acad Sci U S A. 2007 Sep 11;104(37):14825-30. PubMed.
- Bogaert E, d'Ydewalle C, Van Den Bosch L. Amyotrophic lateral sclerosis and excitotoxicity: from pathological mechanism to therapeutic target. CNS Neurol Disord Drug Targets. 2010 Jul;9(3):297-304. PubMed.
- Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, Myszczynska MA, Higginbottom A, Walsh MJ, Whitworth AJ, Kaspar BK, Meyer K, Shaw PJ, Grierson AJ, De Vos KJ. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016 Aug 1;35(15):1656-76. Epub 2016 Jun 22 PubMed.
- Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, Hu F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun. 2016 May 18;4(1):51. PubMed.
- Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM, Everett L, Lenk GM, McKenna-Yasek DM, Weisman LS, Figlewicz D, Brown RH, Meisler MH. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009 Jan;84(1):85-8. PubMed.
Further Reading
News
- Stressed-Out Cells Translate C9ORF72 Repeats, Unleash Toxic Peptides
- Stabilizing C9ORF72 RNA Lowers Toxicity
- C9ORF72 Throws a Wrench into DNA Repair Machinery
- Not Just for Proteins—Expanded RNA Repeats Form Gels, Too
- Antisense RNA from C9ORF72 Repeats Is Likely Culprit in Patient Neurons
- Grow that Axon: C9ORF72 Function Revealed?
- C9ORF72 Knockout Causes Inflammation, not Neurodegeneration
Primary Papers
- Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, Hendricks E, Linares GR, Wang Y, Son EY, Wen X, Kisler K, Wilkinson B, Menendez L, Sugawara T, Woolwine P, Huang M, Cowan MJ, Ge B, Koutsodendris N, Sandor KP, Komberg J, Vangoor VR, Senthilkumar K, Hennes V, Seah C, Nelson AR, Cheng TY, Lee SJ, August PR, Chen JA, Wisniewski N, Hanson-Smith V, Belgard TG, Zhang A, Coba M, Grunseich C, Ward ME, van den Berg LH, Pasterkamp RJ, Trotti D, Zlokovic BV, Ichida JK. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018 Mar;24(3):313-325. Epub 2018 Feb 5 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Director of Neuromuscular Medicine, Cedars-Sinai Medical Center
I think this is a great paper, and makes a convincing argument that haploinsufficiency of C9orf72 can be detrimental to the function of neurons, in addition to similar findings previously shown in myeloid cells. I think it adds further evidence that the disease cannot be simply looked at from the perspective of one potential toxic mechanism (RNA granules, dipeptide repeats) and rather should be looked at as resulting from a combination of gain- and loss-of-function effects within and across cell types in the nervous system.
Yale University
It has long been known that the hexanucleotide expansions in a non-coding region of the C9ORF72 gene that cause ALS and FTD are accompanied by a reduction in C9ORF72 expression (van Blitterswijk et al., 2015; DeJesus-Hernandez et al., 2011; Gijselinck et al., 2012; Belzil et al., 2013). However, while considerable attention has focused on toxic gain-of function pathogenic mechanisms directly arising from the repeat expansions, the contributions of partial loss of the normal C9ORF72 protein to disease have been difficult to pin down. For example, C9ORF72 knockout mice do not develop ALS or FTD-like neurodegenerative phenotypes (O'Rourke et al., 2016; Burberry et al., 2016; Atanasio et al., 2016; Koppers et al., 2015). However, it has been unclear whether this reflects a limitation of the model system or a genuine lack of contribution of C9ORF72 protein deficiency to disease mechanisms.
This new paper from Shi et al. builds on the extensive use of human iPSC-derived motor neurons and data derived from them to propose the provocative new idea that endosome and lysosome dysfunction arising from C9ORF72 haploinsufficiency renders neurons more sensitive to glutamate excitotoxicity due to defects in glutamate receptor traffic. Interestingly, they also found that inhibition of PIKFYVE, a lipid kinase of the endolysosomal pathway, protects C9ORF72-deficient neurons from glutamate-induced excitotoxicity.
These observations refocus attention on the possible disease relevance of C9ORF72 haploinsufficiency and draw attention to the fact that even though recent years have brought many new insights into possible functions for the C9ORF72 protein, there is still considerable uncertainty and a lack of consensus concerning the direct biochemical function or functions of C9ORF72.
Shi and colleagues present evidence that C9ORF72 localizes to and presumably functions on early endosomes but that late endosomes and lysosomes are abnormal in neurons lacking C9ORF72. These findings parallel the lysosome defects that have previously been observed in selected hematopoietic cell types in C9ORF72 KO mice and a human C9ORF72 KO cell line (O'Rourke et al., 2016; Sullivan et al., 2016; Amick et al., 2016). As early endosomes and lysosomes are connected to one another by membrane traffic along the endocytic pathway, it is reasonable to expect that endosomal functions of C9ORF72 could initiate a cascade of changes that ultimately affects lysosomes.
As the field currently stands, there is increasing confidence that C9ORF72 has a function that is somehow relevant to the endolysosomal pathway, but there is also considerable confusion about its precise direct function or functions. For example, in contrast to the neuronal early endosome localization for C9ORF72 that was observed by Shi et al., C9ORF72 was found to very specifically and selectively localize to late endosomes and lysosomes in non-neuronal human cells (Amick et al., 2016). Likewise, in contrast to the early endosome/Rab5 focus presented in the new study, C9ORF72 has also been proposed to act as a guanine nucleotide exchange factor for Rab8a and Rab39b (Sellier et al., 2016; Yang et al., 2016), small GTPases of recycling endosomes and the Golgi (Amick and Ferguson, 2017; Ang et al., 2003; Giannandrea et al., 2010). Interactions between C9ORF72 and Rab1a (Webster et al., 2016), a GTPase with well-characterized functions at the endoplasmic reticulum (Tisdale et al., 1992) have also been reported. Beyond Rab-dependent membrane trafficking functions, C9ORF72 localization to the nuclear envelope via interactions with nuclear pore proteins has also been reported (Xiao et al., 2015). Meanwhile, interactions with the ULK1 complex have suggested a role in the regulation of autophagy (Sullivan et al., 2016; Sellier et al., 2016; Webster et al., 2016).
It has been exciting to see this flood of new insights. However, there is a requirement for greater integration between cell biological, genetic and biochemical approaches to decipher which of these observations are most physiologically relevant and directly connected to the fundamental function of C9ORF72 and the larger SMCR8- and WDR41-containing protein complex in which it resides. Identifying direct functions for the C9ORF72 protein could also help to identify common vulnerabilities that are shared by neurons carrying mutations in the growing list of endolysosomal genes whose mutations cause ALS-FTD that is accompanied by TDP-43 pathology.
An additional finding reported by Shi et al. that is certain to stimulate discussion is the observation that inhibiting PIKFYVE, a lipid kinase of the endolysosomal pathway, protects C9ORF72-deficient neurons from glutamate excitotoxicity. As there is a long history of investigation of glutamate excitotoxicity as an ALS disease mechanism, it would be interesting to know how riluzole or similar drugs compare to PIKFYVE inhibition in the protection of C9ORF72 mutant neurons from glutamate.
The neuroprotective effects of PIKFYVE inhibition are somewhat surprising given that PIKFYVE inhibition interferes with lysosome function by blocking the delivery and maturation of newly made lysosomal hydrolases (Gayle et al., 2017). On the surface, this runs counter to a widespread belief that enhancing lysosome function should be neuroprotective. Likewise, loss-of-function mutations in FIG4, a key binding partner of PIKFYVE, actually cause severe neurodegeneration in mice (Chow et al., 2007) and ALS in humans (Chow et al., 2009). Nonetheless, the PIKFYVE inhibitor known as apilimod was found to be well-tolerated in human clinical trials and is currently under investigation in humans for the treatment of non-Hodgkin lymphoma (Gayle et al., 2017). Therefore, there is precedent for therapeutic efforts to target this pathway in the context of human disease.
While the idea that PIKFYVE inhibition might have therapeutic benefits through modulation of glutamate receptor trafficking is very intriguing, significant follow-up is required to determine whether the beneficial effects observed by Shi et al. in relatively acute assays in very specific models can translate into significant therapeutic benefits in human disease that would exceed those seen with existing glutamate blockers such as riluzole. A major challenge for addressing this issue is the difficulty in faithfully modeling the full range of human ALS-FTD pathology in either transgenic mouse models or human iPSC-derived neurons. Further progress in this area is required to understand the relative contributions of RNA, dipeptide repeat protein, and C9ORF72 protein haploinsufficiency-based mechanisms to ALS-FTD pathology and the ability of candidate therapies to reverse the most deleterious defects.
References:
van Blitterswijk M, Gendron TF, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Daughrity LM, Murray ME, Heckman MG, Jiang J, Lagier-Tourenne C, Edbauer D, Cleveland DW, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Petrucelli L, Boeve BF, Graff-Radford NR, Boylan KB, Dickson DW, Rademakers R. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 2015 Dec;130(6):863-76. Epub 2015 Oct 5 PubMed.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011 Oct 20;72(2):245-56. Epub 2011 Sep 21 PubMed.
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, De Jonghe P, Vandenberghe R, Santens P, De Bleecker J, Maes G, Bäumer V, Dillen L, Joris G, Cuijt I, Corsmit E, Elinck E, Van Dongen J, Vermeulen S, Van den Broeck M, Vaerenberg C, Mattheijssens M, Peeters K, Robberecht W, Cras P, Martin JJ, De Deyn PP, Cruts M, Van Broeckhoven C. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012 Jan;11(1):54-65. PubMed.
Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK, Pregent L, Daughrity L, Baker MC, Rademakers R, Boylan K, Patel TC, Dickson DW, Petrucelli L. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013 Dec;126(6):895-905. Epub 2013 Oct 29 PubMed.
O'Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J, Kim KJ, Bell S, Harms MB, Miller TM, Dangler CA, Underhill DM, Goodridge HS, Lutz CM, Baloh RH. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016 Mar 18;351(6279):1324-9. PubMed.
Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, Suzuki-Uematsu S, Ghosh S, Singh A, Merkle FT, Koszka K, Li QZ, Zon L, Rossi DJ, Trowbridge JJ, Notarangelo LD, Eggan K. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016 Jul 13;8(347):347ra93. PubMed.
Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, Fury W, Burfeind P, Zamfirova R, Warshaw G, Orengo J, Oyejide A, Fralish M, Auerbach W, Poueymirou W, Freudenberg J, Gong G, Zambrowicz B, Valenzuela D, Yancopoulos G, Murphy A, Thurston G, Lai KM. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016 Mar 16;6:23204. PubMed.
Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sá R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH, van den Berg LH, Pasterkamp RJ. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015 Sep;78(3):426-38. Epub 2015 Jul 3 PubMed.
Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, Hu F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun. 2016 May 18;4(1):51. PubMed.
Amick J, Roczniak-Ferguson A, Ferguson SM. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell. 2016 Oct 15;27(20):3040-3051. Epub 2016 Aug 24 PubMed.
Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, Oulad-Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, Charlet-Berguerand N. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016 Jun 15;35(12):1276-97. Epub 2016 Apr 21 PubMed.
Yang M, Liang C, Swaminathan K, Herrlinger S, Lai F, Shiekhattar R, Chen JF. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv. 2016 Sep;2(9):e1601167. Epub 2016 Sep 2 PubMed.
Amick J, Ferguson SM. C9orf72: At the intersection of lysosome cell biology and neurodegenerative disease. Traffic. 2017 May;18(5):267-276. Epub 2017 Mar 23 PubMed.
Ang AL, Fölsch H, Koivisto UM, Pypaert M, Mellman I. The Rab8 GTPase selectively regulates AP-1B-dependent basolateral transport in polarized Madin-Darby canine kidney cells. J Cell Biol. 2003 Oct 27;163(2):339-50. PubMed.
Giannandrea M, Bianchi V, Mignogna ML, Sirri A, Carrabino S, D'Elia E, Vecellio M, Russo S, Cogliati F, Larizza L, Ropers HH, Tzschach A, Kalscheuer V, Oehl-Jaschkowitz B, Skinner C, Schwartz CE, Gecz J, Van Esch H, Raynaud M, Chelly J, de Brouwer AP, Toniolo D, D'Adamo P. Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am J Hum Genet. 2010 Feb 12;86(2):185-95. PubMed.
Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, Myszczynska MA, Higginbottom A, Walsh MJ, Whitworth AJ, Kaspar BK, Meyer K, Shaw PJ, Grierson AJ, De Vos KJ. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016 Aug 1;35(15):1656-76. Epub 2016 Jun 22 PubMed.
Tisdale EJ, Bourne JR, Khosravi-Far R, Der CJ, Balch WE. GTP-binding mutants of rab1 and rab2 are potent inhibitors of vesicular transport from the endoplasmic reticulum to the Golgi complex. J Cell Biol. 1992 Nov;119(4):749-61. PubMed.
Xiao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, Keith J, Zinman L, Rogaeva E, Robertson J. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol. 2015 Oct;78(4):568-83. Epub 2015 Aug 29 PubMed.
Gayle S, Landrette S, Beeharry N, Conrad C, Hernandez M, Beckett P, Ferguson SM, Mandelkern T, Zheng M, Xu T, Rothberg J, Lichenstein H. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood. 2017 Mar 30;129(13):1768-1778. Epub 2017 Jan 19 PubMed.
Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, Szigeti K, Shy ME, Li J, Zhang X, Lupski JR, Weisman LS, Meisler MH. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007 Jul 5;448(7149):68-72. Epub 2007 Jun 17 PubMed.
Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM, Everett L, Lenk GM, McKenna-Yasek DM, Weisman LS, Figlewicz D, Brown RH, Meisler MH. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009 Jan;84(1):85-8. PubMed.
Cornell University
This study clearly demonstrates that C9ORF72 haploinsufficiency plays a role in disease and that likely all three mechanisms—C9 haploinsufficiency, RNA repeat toxicity, and dipeptide repeat toxicity—contribute to the disease progression. It is interesting that both isoforms of C9ORF72 can rescue motor neuron survival and that inhibiting PI(3,5)P2 synthesis can rescue the phenotype as well. In addition, they show that C9ORF72 is localized to early endosomes and C9 deficiency leads to defects in lysosomal biogenesis. This indicates endolysosomal system could be a novel therapeutic target for C9-ALS.
University of Toronto
University of Toronto
University of Toronto
University of Toronto
The paper by Shi et al. indicates the significance of normal function of C9ORF72 and its involvement in the lysosome/autophagosome pathway. This study reports that neurodegeneration in C9ORF72 carriers could be a result of the combined action of gain-of-function and loss-of-function mechanisms.
One of the key implications of this study is that increasing C9ORF72 activity in motor neurons could mitigate disease. Notably, our recent study is in agreement with such a possibility (McGoldrick et al., 2018). We reported a mosaic C9ORF72 carrier with upregulated C9ORF72 expression, in which the presence of RNA foci and DPR pathology was insufficient for disease manifestation and TDP-43 pathology. Specifically, we investigated a neurologically normal individual at age 90 carrying a 70-repeat C9ORF72 allele in blood that expanded to a pathogenic range in offspring (~1,750 repeats). The 70-repeat allele is not methylated (in contrast to DNA methylation of large expansions leading to transcriptional silencing of C9ORF72 (Xi et al., 2015). Remarkably, RNA foci and DPR burdens were similar or even significantly increased in the unaffected father compared to the daughter with ALS. However, the father did not display TDP-43 pathology or signs of neurodegeneration. His CNS tissues had different degrees of mosaicism of small (70-repeat) and large (>1,000 repeats) expansions. Surprisingly, in each mosaic tissue, C9ORF72 mRNA levels were significantly increased compared to an ALS-affected daughter (e.g., by 51 percent in cerebellum, 45 percent in frontal cortex, 325 percent in cervical spinal cord, and 106 percent in thoracic spinal cord). Increased C9ORF72 expression correlated with higher levels of the 70-repeat allele (McGoldrick et al., 2018).
Of interest, the highest C9ORF72 expression among CNS tissues of unaffected individuals was detected in the cerebellum (Renton et al., 2011) with high abundance of DPRs in C9ORF72 cases but no degeneration (Mackenzie et al., 2015).
Small expansions (e.g., 70 repeats) might be considered ‘‘pre-mutations’’ to reflect their propensity to expand in the next generation. Caution should be taken when consulting carriers of small expansions because disease manifestation could be dependent on the extent of the somatic instability in disease-relevant tissues (associated with different levels of C9ORF72 expression). Follow-up studies might help explain the high frequency of ALS- or FTD-affected individuals with an expansion but without a familial history (e.g., 21 percent among Finnish ALS subjects).
References:
McGoldrick P, Zhang M, van Blitterswijk M, Sato C, Moreno D, Xiao S, Zhang AB, McKeever PM, Weichert A, Schneider R, Keith J, Petrucelli L, Rademakers R, Zinman L, Robertson J, Rogaeva E. Unaffected mosaic C9orf72 case: RNA foci, dipeptide proteins, but upregulated C9orf72 expression. Neurology. 2018 Jan 23;90(4):e323-e331. Epub 2017 Dec 27 PubMed.
Xi Z, Zhang M, Bruni AC, Maletta RG, Colao R, Fratta P, Polke JM, Sweeney MG, Mudanohwo E, Nacmias B, Sorbi S, Tartaglia MC, Rainero I, Rubino E, Pinessi L, Galimberti D, Surace EI, McGoldrick P, McKeever P, Moreno D, Sato C, Liang Y, Keith J, Zinman L, Robertson J, Rogaeva E. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. 2015 May;129(5):715-27. Epub 2015 Feb 26 PubMed.
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chiò A, Restagno G, Borghero G, Sabatelli M, ITALSGEN Consortium, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011 Oct 20;72(2):257-68. Epub 2011 Sep 21 PubMed.
Mackenzie IR, Frick P, Grässer FA, Gendron TF, Petrucelli L, Cashman NR, Edbauer D, Kremmer E, Prudlo J, Troost D, Neumann M. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015 Dec;130(6):845-61. Epub 2015 Sep 15 PubMed.
Make a Comment
To make a comment you must login or register.