2019—A Year of Hope for Alzheimer's Research
Quick Links
In the year just past, Alzheimer’s researchers, families, and stakeholders felt renewed hope that new treatments might be within grasp. While the Lazarus story of aducanumab may or may not be enough for FDA approval this year, data from its Phase 3 program solidified a broader signal across four different anti-amyloid antibodies that amyloid can be removed from the brain and that maybe—just maybe—this will also benefit cognition and function if given early at a sufficient dose. The prospect that the amyloid hypothesis is druggable, alone, was enough to re-energize the field. The hope that further trials to define the best doses, patient groups, and treatment regimens will eventually pay off was cause for even more enthusiasm.
A boost in funding announced as the U.S. Congress headed for its holiday break also gave cause for celebration going into 2020, though the funding picture is less rosy in other countries. The NIH budget for AD research now stands at $2.8 billion, a $350 million increase over 2019.
How to spend it? The range of research in the field these days is truly broad. If 2019 trends continue, we can expect to see machine learning brought to bear on more big datasets—for genomics, proteomics, lipidomics, metabolomics, and indeed epidemiology and health economics questions in Alzheimer’s research. Biomarker research is expanding on multiple fronts. New markers are being developed for synaptic and other aspects of neurodegenerative disease, CSF markers are entering clinical practice in some places, and blood tests are entering clinical trials. iPSC-based analysis of patient neurons and glia is coming into its own, revealing new aspects of molecular pathogenesis. Single-cell transcriptomics to tease out exactly how specific cell types change over the course of disease, or even how specific cells near a plaque respond, holds out hope that scientists may finally find an explanation for why certain brain regions are particularly vulnerable to one neurodegenerative disease or another. And then there’s microglia. It seemed you couldn’t swing a pipette without hitting a cluster of these immune cells in 2019, or open a journal without reading about them.
PubMed listed 12,000 papers on Alzheimer’s disease in 2019, and 800 on frontotemporal dementia (FTD). Peruse below our best attempt to wrangle the main advances—and write to us about what we forgot.
1. Clinical Trials
From down and out in spring to back up in the fall, 2019 was a wild ride for aducanumab. The disappointment was great when, in March, Biogen halted the Phase 3 ENGAGE and EMERGE trials of this therapeutic antibody, because a futility analysis indicated no slowing of cognitive decline (Mar news). In October Biogen reversed itself, saying further analysis of a larger dataset now suggested aducanumab worked in those participants who had received 14 injections of the highest dose (Oct news).
What gives? Midway through the trials, Biogen had introduced protocol changes. One allowed ApoE4 carriers to now receive the highest dose of 10 mg/kg, whereas initially they had been capped at 6 mg/kg over concerns about ARIA; another allowed ApoE4 carriers to resume their original aducanumab dose after their ARIA had resolved. Essentially, these changes increased drug exposure over time in some of the later enrollees, but not earlier enrollees, especially in EMERGE, which started after ENGAGE did. In the former, Biogen reported a slowing of cognitive decline by a quarter and functional decline by 40 percent in people on the highest dose. This was not seen in ENGAGE, however.
At the Clinical Trials on Alzheimer’s Disease (CTAD) conference in November, Biogen reported a widely debated post hoc subgroup analysis, claiming aducanumab had slowed cognitive decline by 27 percent in those ENGAGE participants who did receive the maximum dose (Dec conference news and commentary).
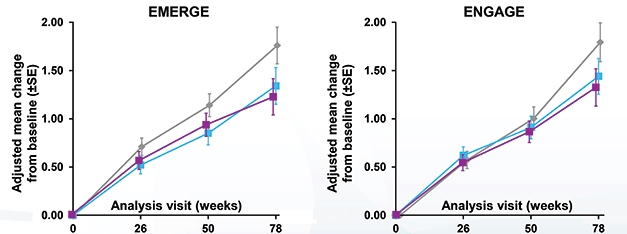
Was Exposure the Key? A post hoc subgroup analysis of participants enrolled after maximum dosing was raised claimed there was a cognitive benefit in both trials (gray, placebo; purple, high dose). [Courtesy of Biogen.]
Imaging data from florbetapir amyloid and MK640 tau PET sub-studies confirmed that aducanumab lowered brain amyloid, and may have even reduced neurofibrillary tangles a tad.
Leading researchers generally believed the overall signal seen with aducanumab. It revived their enthusiasm for anti-amyloid immunotherapy and bolstered the notion that the amyloid hypothesis is druggable. That said, they wanted to see more data and questioned Biogen’s plan to seek regulatory approval based on the current dataset.
Other Aβ immunotherapies, especially the antibodies BAN2401, gantenerumab, and donanemab, all appear to push brain amyloid below the threshold of positivity, as well, and possibly even keep it there for a few years after antibody treatment is stopped (see image below). Scientists uniformly welcomed the current trend of calling immunotherapy trial participants back for open-label extensions to watch how their amyloid load, disease progression, and potential side effects unfold over several years. By contrast, crenezumab removed no plaques, but it did nudge down neurogranin and other neuro-inflammatory and injury markers (Dec conference news).

Different Trajectories, Same Result. Three representative patients in the gantenerumab long-term extension study cleared plaque at different rates, but all ended up below zero at year three. [Courtesy of Roche.]
On the down side, Roche shelved crenezumab (Jan news). Interim analyses of the Phase 3 CREAD1 and CREAD2 trials indicated neither was likely to reach their primary endpoint of slowing cognitive decline in people with prodromal or mild AD. Crenezumab primarily binds soluble Aβ oligomers. A prevention trial in people carrying the Paisa presenilin 1 mutation that causes autosomal-dominant AD is continuing.
The active anti-Aβ immunotherapy CAD106 also ran aground in 2019. Novartis suspended its dosing in the Generation Phase 2/3 program to enable unblinded interim analyses of the halted BACE inhibitor umibecestat, which was being jointly evaluated with CAD106 in the same trial (see page 18 of Novartis financial report).
As a group, BACE inhibitors were battered last year. In April, Merck formally published results of its Phase 3 trial of verubecestat in prodromal AD, where it had worsened cognition (Apr news). The next blow came in July, when Novartis/Amgen halted the two Generation trials of umibecestat in cognitively normal people at high risk for AD. This inhibitor, too, slightly worsened scores on the RBANS composite, and accelerated brain atrophy (Jul news). Months later, Biogen/Eisai halted their Phase 3 Mission program of elenbecestat, citing similar concerns (Sep news). Umi- and elenbecestat are about threefold selective for BACE1 over BACE2, putting paid to the idea that this would solve the problems of the earlier, nonselective BACE1/2 inhibitors.
Still, researchers refuse to give up on BACE inhibitors. The dramatic inhibition in these trials may be too much, too late, the thinking goes, and lower doses may yet work in a prevention paradigm. Others argue for a better understanding of these cognitive changes. They seem to affect mostly memory, occur in the first week or two after dosing begins, and then stabilize (Dec 2019 conference news and commentary).
In 2019 the first tau immunotherapy results appeared, and they were not good. In July, AbbVie halted its Phase 2 trial of ABBV-8E12 in progressive supranuclear palsy (PSP), a primary tauopathy, after an interim futility analysis (Jul news). AbbVie scrapped two extension trials in PSP, and plans for trials of other primary tauopathies, but is continuing a trial in AD. Next to fall was gosuranemab, aka BIIB092. This antibody hit its target in Phase 1, reducing extracellular N-terminal tau fragments in CSF; alas, no difference emerged between placebo and treatment groups on the PSP rating scale in the Phase 2 PASSPORT trial (May news). In December Biogen halted PASSPORT, and also TauBasket, a trial to test gosuranemab in four other primary tauopathies (Dec news).
It’s early days for tau therapies. Researchers don’t automatically assume that an antibody that flopped in primary tauopathies will fail in AD, as well. In part that is because neurofibrillary tangles in AD comprise two different isoforms of tau containing either three or four repeats at the C terminus, whereas in primary tauopathies four-repeat tau tangles predominate. AbbVie, Biogen, Janssen, Lilly, Genentech, and other companies are developing tau immunotherapies, tau RNA-based therapies, and other tau-based approaches in AD trials.
2019 brought ups and down for anti-inflammatory strategies. The coffin finally closed on the idea that nonsteroidal anti-inflammatory drugs can prevent Alzheimer’s disease, when the INTREPAD trial failed (Apr news). Two years of naproxen in asymptomatic people at high risk for AD made no difference on cognition, structural or functional brain imaging, or CSF Aβ, total or phospho-tau, all the while causing significant gastrointestinal and cardiovascular side effects.
The epigenetic drug apabetalone, on the other hand, may quell brain inflammation indirectly, by acting not in the brain but in the blood. By blocking transcriptional machinery, this drug shuts down hundreds of genes, including pro-inflammatory cytokines made by endothelial cells (see image below). It is being tested primarily in cardiovascular disease, but in a cognitive sub-study seemed to help people with mild cognitive impairment or early dementia. Likewise, Grifols’ plasma exchange and albumin replacement therapy (AMBAR) is said to remove Aβ from the brain by pulling Aβ protein complexes from the blood. The Barcelona-based blood products company reported positive results from its Phase 2b/3 study at CTAD. AMBAR was reported to slow cognitive and clinical decline over 14 months, though questions about the study protocol curbed enthusiasm among clinicians (Dec conference news).

Mechanism of Action. Bromodomain and extra-terminal (BET) proteins bind acetylated lysines (ac) in histones and recruit transcription factors (TF); apabetalone (yellow) competes with these modified lysines, preventing transcription. [Courtesy of Ewelina Kulikowski.]
Others are pinning hopes on new ways to remove Aβ from within the brain. Alector and Denali, both in South San Francisco, are developing antibodies that activate TREM2 and are intended to spur microglia to clear amyloid (see image below and May conference news). Alector’s AL002 started Phase 1 in November 2018 and AL003, an antibody that suppresses CD33, which tempers TREM2 signaling, started in March 2019 (Apr conference news).

TREM2 Activation. Binding an antibody (red) to TREM2 can trigger signaling through its co-receptor DAP12, resulting in phosphorylation of Syk. [Courtesy of Donna Wilcock.]
Last year brought some good news for disease management. Acadia Pharmaceuticals’ Phase 3 trial for the selective serotonin inverse agonist pimavanserin wrapped up early when the drug reached its primary endpoint of delaying relapse to hallucinations and delusions in people with dementia (Sep news). This study used an unusual design, testing how well patients did when they came off pimavanserin rather than when they went on it. After a 12-week open-label phase, patients who responded to the drug either continued it or were switched to placebo. At CTAD, Acadia reported that 75 percent of patients responded to the drug in the open-label phase of the trial. Among those who continued on to the blinded portion, the drug delayed relapse by 65 percent. Those randomized to the drug were three times less likely to relapse than were those switched to placebo.
In China, regulators conditionally approved GV-971 for the treatment of Alzheimer’s. This drug was met met with skepticism but went on sale on December 30 (see South China Morning Post news; PubPeer). A mixture of oligosaccharides extracted from kelp, GV 971 reportedly slowed cognitive decline in a Phase 3 trial. Unusual cognitive trajectories in placebo and treatment arms prompted scientists to call for formal publication of the Phase 3 results. An international trial is being planned (Nov news).
In 2019, interest in gene-based therapies to treat AD and related dementias exploded. This was driven in part by the success of gene therapy in spinal muscular atrophy. In October, a Phase 1 trial in AD patients homozygous for ApoE4 started; it injects an AAV-APOE2 construct designed to counteract the effects of ApoE4 into the brain (Nov conference news).
Beyond AD, manifold gene therapy approaches into lysosomal storage diseases are taking hold. They may serve as stepping-stones for similar work in AD, where aspects of lysosomal degradation increasingly draw interest as targets (Dec news). After all, scientists made “Milasen,” a personalized oligonucleotide drug to halt progression of a lysosomal storage disorder with features of Parkinson’s and dementia caused by a mutation in the CNL7 gene (Kim et al., 2019; Oct STAT news).
Contrary to public perception, therapeutic approaches in Alzheimer’s disease have for years been much broader than merely targeting amyloid, and 2019 was no different. To cite but two examples, the past year saw innovation in the form of NDX-1017, a small molecule that induces the neuroprotective HGF/MET receptor, and gemfibrozil, a cheap old fibrate drug that activates the PPARα nuclear receptor. Both managed respectable results in well-designed Phase 1 trials and are moving forward (Dec 2019 conference news).
On the clinical trials front, the next big thing—and arguably the most ambitious goal—is primary prevention in people who have no or little amyloid pathology yet. The DIAN project of families with autosomal-dominant AD is leading the way, preparing to start up a trial in mutation carriers in their 20s and 30s in 2020 (Aug conference news).
But still, there is no effective treatment, and supplement companies have rushed in to fill the void, some peddling products with spurious claims. In spring the FDA cracked down, warning 17 companies to stop advertising dietary supplements as AD treatments (Mar news).
2. Genetics
Last spring, three massive genome-wide association studies formally appeared in print (see image below and March news). Those tantalizing but cryptic lists of variants have sparked intense sleuthing among geneticists to nab the variants that underlie the associations, and those efforts are the story of 2019.

Top Hits. A biological ranking system prioritizes genes near known and newly discovered GWAS hits. [Courtesy of Kunkle et al., Nature Genetics, 2019.]
Scientists are combining genetics, genomics, transcriptomics, and expression network analyses to pin down the functional effects of risk variants. Time and again, they find genes expressed primarily in microglia. For example, using data from projects such as ENCODE, researchers mapped known GWAS variants in enhancer regions to gene expression in myeloid cells, uncovering nine genes not previously linked to AD (Aug news). Lo and behold, many of those enhancers bind the master microglial transcriptional regulator PU.1, implying coordinated expression of AD-related genes in these cells. Other groups create their own atlases of active regulatory regions specific to human neurons, astrocytes, oligodendrocytes, and microglia. They too, see AD GWAS hits cluster in regulatory regions of the microglial genome (see image below and click microscope icon for largest view; Nov news).

Hello Again, Microglia. Alzheimer’s risk genes expressed in microglia, which have been identified in one (gray), two (green), or three (yellow) GWAS studies, encode proteins that form an interaction network. [Courtesy of Nott et al., Science/AAAS.]
Many microglial gene variants that increase the risk of AD were found to heighten activation of these cells. This happens after amyloid has begun to accumulate but before any signs of neurofibrillary tangles (Feb news). One protective microglial variant, in the gene for phospholipase Cg2, protects against other dementias, as well (May news; Friedman et al., 2018).
So-called “dark” regions of the genome apparently hide AD variants. Data from the Alzheimer’s Disease Sequencing Project revealed 36,000 DNA segments that sequencing studies typically miss (May news). These were interspersed among 6,000 genes, of which 76 are linked to some 300 diseases. A frameshift mutation in a dark region of a known AD gene, CR1, turned up in five AD cases but no controls among 13,000 ADSP samples, suggesting the frameshift could be pathogenic.

CircRNA in AD? This circular Manhattan plot shows numerous circRNAs (red asterisks) that significantly correlate with dementia severity, Braak score, and/or AD status. Outer rings show chromosomes. Inner three circles show positions of the circRNAs in the genome (alternating light and dark gray), while their radii reflect the strength of the association with the three traits. Dotted gray lines indicate transcripts that link to two of these traits; solid gray lines, transcripts linked to all three. [Courtesy of Dube et al., Nature Neuroscience.]
Even more mysterious, in October scientists told of circles of RNA in the AD brain. Formed when the ends of transcripts get spliced together, such circles are found throughout the eukaryotic world. Some regulate gene expression. In two postmortem cohorts, 164 circRNAs correlated either with AD diagnosis, CDR score, or Braak stage (see image at right). Five circRNAs correlated with all three, including circular transcripts of known AD genes Homer1 and Picalm (Oct news).
In terms of specific genes, 2019 saw a resurgence of interest in ApoE. A large neuropathology-controlled sample showed just how strongly ApoE2 protects. People with two copies of this rare allele have but one-tenth the chance of getting Alzheimer’s as someone with the most common E3/E3 genotype (Reiman et al., 2019). And variants beyond the three main alleles are drawing interest, as well. A Colombian woman with the previously discovered Christchurch ApoE variant has only subtle short-term memory loss in her 70s despite also carrying the Paisa presenilin mutation, which brings on dementia in midlife in almost every carrier. While this septuagenarian has an exceptionally high amyloid load in her brain, she had minimal neurofibrillary tangles (see image below and Nov news). More broadly, a genomic analysis of the larger ApoE locus assigned AD risk to a host of noncoding variants (Zhou et al., 2019).

Plaques but No Tangles. A woman with a familial AD mutation and two ApoE3 Christchurch alleles (left scans) had a very high plaque (top, red) but low tangle burden (middle) and healthy brain metabolism (bottom) compared with a typical mutation carrier (right scans). [Courtesy of Arboleda-Velasquez et al., Nature Medicine.]
Beyond APOE, in May rare variants in APOB, which encodes low-density lipoprotein, were linked to early onset AD (May news). Together, this work is spurring a new round of drug discovery work targeting ApoE and lipid metabolism in the brain.
There is renewed interest in familial AD mutations in APP and PS1, as well. These families carry deterministic mutations, but the 50-year range in their age of symptom onset is influenced by additional risk and protective variants, hence they inform research into the overall genetic architecture of AD (Aug conference news). New pathogenic mutations in APP and PS1 continue to be found, and old mutations inspire mechanistic studies on what exactly they do to the cells expressing them. For example, some were reported to cripple endosomal trafficking in a panel of patient-derived cell lines. Curiously, the endosomal abnormalities were caused by APP C-terminal fragments, not Aβ (Aug news). A neuronal gene that rose in prominence across AD genetics is Sorl1, which encodes a receptor involved in sorting and processing APP. This huge gene harbors a great variety of common SNPs as well as rare high-risk missense and truncating variants (Campion et al, 2019).
Can the rapidly growing list of AD variants be computed into a single number that tells a person his or her risk? Not quite yet, but researchers are working on it. In 2019, a polygene risk score correlated with the presence of plaques, tangles, cortical atrophy, and cognitive decline, beating ApoE or imaging for predictive power (Feb news). The hope is it will make a good screening tool soon.
3. Pathogenesis/Molecular Genetics
In this research area, 2019 was the year of the microglia. Looking to specific microglial genes, researchers got a better handle on how variants in ApoE, TREM2, CD33, Bin1, the MS4A gene cluster, and progranulin either soothe microglia into a homeostatic state, or aggravate them into a cytokine-spewing one (Apr conference news). MS4A4A and TREM2 were spotted cozying up to each other in the cell membrane, where the former drives shedding of the latter, and indeed MS4A variants correlated with levels of the soluble fragment of TREM2 in CSF. Polymorphisms associated with more CSF sTREM2 decrease a person’s chances of AD, while those associated with less sTREM2 increase it (Aug news). Bolstering the idea that TREM2 shedding is protective, people with plaques were sharper if their CSF sTREM2 was high (Aug news).

At the Heart of It All. Many AD gene variants exert their effect in microglia, where they influence the response to amyloidosis. [Courtesy of Annerieke Sierksma.]
Do microglia protect by gobbling up Aβ and tau and preventing their spread? When mice lacked TREM2, amyloid plaques grew wider (Jan news), and AD brain extracts more aggressively seeded neurofibrillary tangles, which then spread more readily across the brain (Jul conference news).
Mechanistically, ApoE4, too, was front and center in 2019. Alas, findings thus far are so complicated and sometimes contradictory that a clear story has not yet emerged. Over the last few years it became apparent that ApoE and TREM2 coordinate microglial responses in AD, switching them from homeostasis into a neurodegenerative state (Sep 2017 news; Feb 2015 conference news). Scientists learned that ApoE4 locks microglia into a homeostatic state, leaving the brain vulnerable to myriad pathologies. Others reported that while ApoE4 squelches microglial phagocytosis, it ramps up their inflammatory responses (May conference news; Sep news).
New Teacher: Transcriptomics
This methodology blossomed in 2019, with more labs adopting single-cell and network analyses to tease out how different cell types behave in Alzheimer’s disease. In mouse models and human data, expression of many genes, including known AD genes, were found to ramp up in response to amyloidosis. Most are microglial and many play a part in inflammation (Apr conference news). Similarly, network analyses revealed coordinated regulation of groups of genes in tauopathy mice. One cadre of neuronal genes was suppressed, while another, comprising inflammatory microglial and astrocyte genes, was activated. Homologous expression changes were found in human tissue from AD, FTD, and ALS patients, suggesting common neurodegenerative expression signatures (Mar news).
Spatial transcriptomics emerged as a way to correlate changes in gene expression at the cellular level with neuropathology, perhaps offering an answer to the old question about why specific regions of the brain are more vulnerable in specific age-related neurodegenerative diseases. This method uncovered an upregulated network of 57 genes around plaques in APP knock-in mice (Aug news). In situ hybridization localized most of these—no surprise—to microglia and astrocytes (see image below). The network includes known AD genes, such as TREM2 and APOE, but also 36 not previously linked to plaques.
![]()
Cell-Specific Expression. In a coronal section of the mouse brain (left), binding of probes for each of 57 plaque-induced genes, and for 27 cell-type-specific transcripts (bottom right), identify transcripts expressed by microglia, oligodendrocytes, astrocytes, and neurons, which are up- or downregulated near plaques (white, top right). [Courtesy of Chen et al., 2019.]
4. Microglia
We are nowhere near done with microglia just yet, despite the 20 mentions already in this story. The year 2019 saw a flourishing of human data in AD research, and much of it supported the role of microglia in pathology. For example, in postmortem brain from Chicago’s ROSMAP study, AD pathology correlated with microglial activation, as judged by how many of those cells assumed an amoeboid shape. These rounded microglia seemed to emerge after plaques had formed but before tangles had (Feb news). Dozens of genetic variants associate with these microglia, and most of them are AD risk genes.

Clustering. Transcriptional profiling to define subtypes of human microglia. Each dot represents one cell, colors denote clusters. C5, C6, and C7 are homeostatic microglia; C4 are partially activated cells; C2, 3, and 8 are in MS lesions. C1, 9, and 10 are non-microglia cell types. [Courtesy of Masuda et al., 2019 Nature.]
Efforts to characterize microglia in the brains of living people got quite exciting in 2019. In February, scientists described seven different types, using transcriptomics. They separated cells from cortical tissue taken from people having surgery to control epilepsy, or from people with multiple sclerosis (MS) who were having biopsies of active lesions. From the 1,602 individual microglia they managed to isolate, single-cell RNA-seq then determined their gene expression profiles (Feb news). The profiles plopped the cells into seven major clusters (see image above). Three clusters were solely in the MS lesions, hence represented a disease phenotype. Three looked homeostatic thanks to high expression of core microglial genes. The fourth was brimming with upregulated inflammatory genes, and occurred in both MS and epilepsy.
In November, this profiling project added biopsy tissue from the cancer-free margins of brain tumor resections and, this time, took anatomy and age into consideration as well. The new round partitioned 6,000 microglia into 14 clusters. Four expressed MHC-II and CD68 antigen-presenting genes. These cells hung out mostly in white matter, whereas most microglia in gray matter were not expressing these genes. Brains from people aged 14–30 had predominantly homeostatic microglia, whereas microglia expressing inflammatory genes were more numerous in people over 50 (Nov news). This 2019 work expanded a previous description of 14 microglial clusters from a different group of epilepsy patients (Olah et al., 2018).
Some human microglia clusters corresponded with clusters seen in healthy mice, while others correlated with microglia found in mice that were treated with cuprizone to simulate MS. This suggests some mouse-human conservation of MS phenotypes; alas, the Alzheimer’s field may not be so lucky.
This spring, one study tied a microglial cluster identified in brain samples from ROSMAP to AD (May news). These cells expressed only 28 of the 229 genes expressed by disease-associated microglia (DAM) previously found to congregate near amyloid plaques in mice (Jun 2017 news). On the flip side, the human AD microglia expressed 49 genes never reported in any mouse microglia. Similarly, human Alzheimer’s microglia pried from frozen tissue looked nothing like the DAMs in mouse amyloidosis models (May news). ApoE was the lone gene upregulated in both.
Instead, scientists saw a signature in human MS tissue that overlapped with mouse DAMs, suggesting that DAM reflect more of a general disease signature than a specific amyloidosis one. Even the homeostatic mouse microglial signature looked little like the human equivalent. All told, 2019 made clear that mouse microglia make a poor model for the human counterpart, which is partly what prompted researchers to characterize microglia from human biopsy tissue. In fact, eight out of 39 AD genes, including the microglial genes CD33 and CR1, have no mouse homolog, and the similarities between another 10, including TREM2, and their mouse versions are weak.
Can science model human microglial dynamics in mice at all, then? A new attempt came in the form of chimeric models, where microglia in young mice are replaced with human microglia (see image below). The human cells took to their new environment, tiling across the brain and responding to injury and inflammation. In 5xFAD mice, human microglia crowded around plaques, where they expressed a different set of genes than did mouse microglia. Human microglia with the R47H TREM2 AD risk variant were sluggish in mobilizing around the mouse plaques (Apr conference news; Aug news).

Chimera. Two months after transplanting progenitors into mouse pups, human P2RY12-positive microglia had dispersed throughout the forebrain. [Courtesy of Morgan Coburn and Mathew Blurton Jones.]
As for handy tools, this year saw the generation of mice with glowing microglia. Using gene editing, the microglia-specific TMEM119 gene was targeted to drive expression of florescent markers (Jun news). These mice will make it easier to track microglia and visualize what they do in neurodegeneration models.

Brain or Blood? In the cortices of 5xFAD mice that lack microglia, amyloid (green) accumulates only in blood vessels, not gray matter (left, low mag; right, high mag). [Courtesy of Spangenberg et al., Nature Communications.]
Still, there is much to learn about microglia from mice. A picture of a fearsomely dynamic response is emerging, where microglia may not always protect against pathology, but contribute to it. For example, in 5xFAD mice, researchers ablated microglia by feeding the animals an antagonist to the microglial survival factor CSF 1. Amyloid deposition slowed in the brain parenchyma (see image above right). It wasn’t gone, though, but instead piled up in blood vessels (Sep news). The few plaques that did form in the parenchyma were diffuse and contained little ApoE. These finding jibed with a report earlier in the year that microglia may “inject” ApoE into plaques (Jan news).
ApoE4 also fuels neurofibrillary tangles in mice, but again, not without microglia there. In P301S-tau/ApoE4 mice, eliminating microglia prevented the spread of tau pathology and spared neurons from degenerating (see image below). Knocking out ApoE equally protected neurons (Oct news).

Deadly Alliance. Tau/ApoE4 mice on control chow (left) had brain atrophy and an enlarged ventricle. Removing microglia prevented atrophy (middle). So did removing ApoE (right). [Courtesy of Shi et al., JEM, 2019.]
Microglial ablation was a topic of renewed debate in 2019, because a decade earlier, a study had eliminated microglia from a more aggressive model of amyloidosis, and that did nothing to plaques (Grathwohl et al., 2009). What gives? Is it speed of disease progression, where microglia make no difference when amyloidosis is fast but do if it’s slow? Or perhaps removing microglia from the brain changes cell population dynamics and crosstalk so much that ablating a cell type in mice cannot prove that plaque formation is a direct effect of microglia.
What, if anything, do microglia ablation experiments say about ApoE and human AD? People, too, accumulate more neurofibrillary tangles if they carry ApoE4, which also seems to hasten tangle-related memory loss (Nov news). Could this explain gender differences in AD? Among people with plaques who are cognitively healthy, women accumulate more tangles in the entorhinal cortex than do men, and ApoE4 exacerbates this sex difference (Feb news). Whether this revolves around microglia remains to be seen.
Let’s not forget astrocytes. They got less attention in 2019, but that may well change again, as a subset was found that regulates spine density and synaptic function in surrounding cortical neurons. Their disruption likely contributes to neurodevelopmental disorders in people (Apr news). Astrocytes also might make ready raw material for therapeutic cellular “conversion” attempts into neurons (Jul conference news).
5. Biomarkers
The prospect of a blood test for Alzheimer’s—arguably the greatest need outside of effective treatments—drew closer in 2019. A clinical validation study showed that a mass spectrometry test for plasma Aβ42/40 can tell if a person has abnormal CSF Aβ42 or abnormal amyloid PET (Aug conference news). The Elecsys plasma immunoassay performed similarly well, though both tests needed to factor in ApoE genotype to push accuracy into the high 80 percentiles (Jun news).
The field is hotly debating whether plasma assays are ready for routine clinical use. In a round-robin study of 11 different mass spec and immunoassays measuring aliquots of the same plasma samples, the assays barely correlated (see image below). While agreement among MS tests was better than among immunoassays, scientists disagreed on what this meant (Aug Q&A).
Different immunoassays of CSF Aβ correlate well, hence it could be that the plasma itself explains the weak correlation among these tests in the round robin. Blood is a more complex mixture than CSF; its Aβ42 concentration is 10-fold lower, and peripheral cells release their own Aβ into blood. Another possibility is that immunoassays detect different Aβ42 conformations in the plasma; this could be important if these tests were to monitor responses to immunotherapy. Inconsistencies across assays—which have to perform near their detection limit to pick up a solute that also rises and falls on the tides of biological parameters such as blood pressure, diabetes, diet, or exercise—prompt leaders in the field to urge a thoughtful stepwise deployment of these tests, first with limited applications such as clinical trials and population-based studies.

Head to Head. Overview of correlations from a plasma Aβ round-robin study. Green denotes tight correlation between any given two tests; red, weak correlation. [Courtesy of Henrik Zetterberg and Kaj Blennow, University of Gothenburg.]
Several plasma tests may come to market in 2020. Their purveyors hold out a promise of cost savings, at least in clinical trials that currently use thousands of amyloid PET scans at screening. Some estimates claim savings of many millions of dollars for a 500-person trial, but then again, prices for these tests have not been formally set yet. A less disputed promise: Blood tests will cut screening times for a trial from years to months (Aug conference news).
Plasma p-Tau: The Super-Early Marker You’ve Always Wanted?
Throughout 2019, there were murmurs that biomarker mavens were even more excited about blood tests for tau than they were about the impending plasma Aβ tests. Why? Some phosphorylated forms of tau could soon rival, if not outperform, plasma Aβ as early markers of AD. P-tau offers a bigger signal, since its concentration climbs 10-fold or more in disease, while Aβ ratios typically change within a 15 percent range. So what’s the scoop?
First hints about this came from the CSF of AD patients, where researchers detected hyperphosphorylation of a slew of different tau residues with high-resolution mass spec (see image below). Two stood out. In cross-sectional samples from the DIAN cohort, higher phospho-serine 217 and phospho-threonine 181 were seen 21 and 19 years before estimated disease onset, respectively. That is close to the onset of amyloidosis. CSF p217-tau correlated with PiB binding in the precuneus, an early depositing region, and predicted amyloid positivity with 97 percent accuracy (Aug conference news).
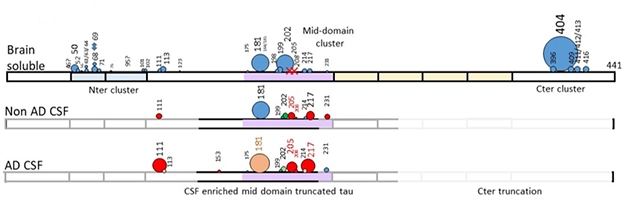
Hot Spots. Degree of phosphorylation (circle diameter) in normal brain, normal CSF, and AD CSF. Orange and red represent slight and high hyperphosphorylation, respectively, compared with normal brain. [Courtesy of Nicholas Barthélemy.]
The same is true in the Swedish BioFinder cohort. CSF p181-tau and p217-tau tracked higher in volunteers who had positive amyloid scans but normal cognition. By contrast, tau PET only detects neurofibrillary tangles once people become impaired. These p-tau values were higher still in cognitively impaired people, and highest in people with AD dementia. Longitudinally, the p-tau markers rose by up to 14 ng/mL over two years in cognitively healthy volunteers. Just as CSF Aβ42 dips a few years before amyloid PET scans turn positive, levels of these p-tau markers change before flortaucipir PET scans turn positive. Alas, in the case of p-tau, the time delay between the fluid tests and current tangle tracers is two decades (Aug conference news). All told, it seems these p-tau species flag Alzheimer’s disease proteopathy very early on indeed.
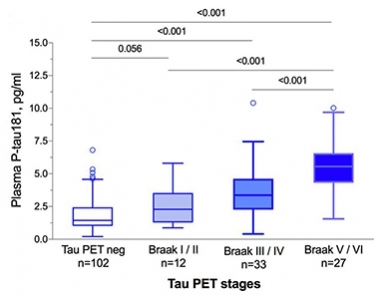
In Synch. p181-tau in the plasma track with Braak staging as determined by tau PET. [ Courtesy of Oskar Hansson.]
What about in the blood? Here’s where AD researchers got truly amped up. In 2018, a first inkling came of an immunoassay picking up p181-tau in plasma and rising with AD severity (Mielke et al., 2018). Would it hold up? Indeed, plasma and CSF p181-tau correlated with each other and with Braak staging in BioFinder (see image at left), and plasma p181-tau was higher in AD than in MCI or in frontotemporal dementia in a Californian cohort. Curiously, when a person’s FTD featured neurofibrillary tangles comprising both three-repeat and four-repeat tau, the type found in AD, then their plasma p181-tau was high (Aug conference news).
Is p181-tau a tangle marker, then, or a harbinger of them? Its appearance many years before tangles suggests the latter, though this gap in time could also reflect higher sensitivities of fluid assays than of PET. Perhaps p181-tau and tangles go hand in hand, but tangles go undetected for longer. Alternatively, p-tau may represent an early stress response to the presence of amyloid, and a sign of future neurodegeneration.
About All These Other Markers …
Aβ and tau are core markers, but they are far from the only ones. Consider neurofilament light chain (NfL), for one. It was confirmed as a general marker of neurodegeneration in 2019. Multivariate analysis showed plasma p181-tau better predicted future AD than did plasma Aβ42/40, total tau, or NfL (Aug conference news). Still, how fast NfL rose in blood better predicted progression than its absolute amount. In DIAN participants, for example, this rate of change accelerated as they neared their age of symptom onset (Jan news).
Beyond Aβ, p-tau, and NfL, other markers may be important. But how to evaluate a rapidly growing slate of candidates? Enter NeuroTool Kit, a trans-Atlantic public/private project that has developed a platform to evaluate fluid biomarkers side-by-side in the same biological specimens from well-defined cohorts (Aug conference news). The idea is to evaluate in a standardized way which markers perform best. Initially, the project conducted 16 tests of 12 markers and four combinations (see image below), but more are to come (Dec conference news).

NeuroTool Kit. Twelve different markers, plus four ratios, were measured side-by-side, in the same way, in the same normal (blue), MCI (yellow), and AD (orange) dementia samples of the Wisconsin ADRC cohort. Each sample is further subdivided as p-tau/Aβ42-negative (left) or -positive (right). [Courtesy of Kaj Blennow.]
Newcomers burst on the scene, too. For example, neural pentraxin emerged as a potential flag for cognitive decline. NPTX2 forms complexes on the neuronal surface that regulate AMPA receptor recycling. Among people diagnosed with MCI or AD, those with the lowest CSF NPTX2 declined quickest on learning and memory tests. The CSF NPTX2/tau ratio better predicted AD than did CSF Aβ42/tau (Aug conference news). NPTX2 also turned up in a proteomics analysis of ROSMAP and BLSA cohorts, where it correlated with cognitive decline and AD, respectively (Aug conference news).
A new CSF assay for a truncated version of synaptic vesicle glycoprotein 2A also drew note. Previously, PET imaging with the ligand UCB-J indicated that SV2A disappears from the brains of people with epilepsy or AD. A fluid marker would be cheaper and, indeed, less of the SV2A fragment turned up in CSF of AD patients than in normal controls (Aug conference news).
And could AD diagnosis be as simple as tracking sleeping patterns? Sleep has long been studied for its importance in memory consolidation and even clearance of Aβ from the brain. Now it appears that reduced slow-wave brain activity during sleep correlates with amyloid load, while weak coupling between slow brain oscillations and an EEG pattern called sleep spindles associated with greater tau burden in the medial temporal lobe (Jun news).
6. Protein Structure
The year 2019 continued a trend in structural biology, in which cryoEM delivered, seemingly at a steady clip, a slew of eye-popping, high-resolution pictures of the core Alzheimer’s and related disease proteins straight from human brain. A surprise twist came with the first structure of Aβ fibrils isolated from people with AD. The protofilaments wrap around each other in a right-handed turn when forming a fibril (see image below and Nov news). Most β-sheet protein filaments twist left, as do tau and synthetic Aβ fibrils. The latter poorly seed amyloidosis compared with fibrils from brain; scientists now think this might be because they twist the wrong way.

Turning Right. Side view of right-handed fibril (left). At right, cross-sectional view of a protofilament, consisting of two C-shaped Aβ40 peptides, positioned back-to-back and stacked (bottom). [Courtesy of Kollmer et al., Nature Communications, 2019.]
CryoEM also caught APP and Notch in the arms of γ-secretase (Jan news). The structures predict that the transmembrane helices of APP unwind before being cleaved. They expose differences between APP and Notch binding that might help drug discoverers develop specific inhibitors of APP processing. Biochemical studies suggested that γ- and β-secretase form a massive complex to process APP (Jan news). If this finding stands, then disrupting this complex might curtail Aβ production without blocking either enzyme directly.
The year brought new cryoEM structures for tau, as well. Tau filaments isolated previously from AD and from Pick’s disease brains looked quite different from each other (Aug 2018 news). How about chronic traumatic encephalopathy? In other words, how broad is the range of tau structures across tauopathies? In March, tau filaments from a former American football player and two former boxers were published.

Tau Polymorphs. Type I (left) and Type II (right) filaments contain the same protofilaments, but different interfaces. [Courtesy of Falcon et al., 2019.]
The normal electron microscope revealed one more (CTE type I) and one less abundant (CTE type II) filament (Mar news). Type I differed from filaments found in Alzheimer’s or Pick’s, but type II was shaped similarly to the paired-helical filaments of AD. At the higher resolution of cryoEM, both type I and II comprised C-shaped protofilaments similar to those in AD. As in AD, they stacked on top of each other back-to-back to form fibrils, but did so in a slightly different orientation (see image above). The C-shape was more open in CTE than in AD. Curiously, an unknown molecule nestled in the CTE filaments (image below). Unmasking this mystery molecule, and understanding more generally what makes tau filaments structurally different, could help explain differences between various tauopathies.

Mystery Molecule. Compared with the AD tau fold (middle), the CTE fold had a more open C-shape, and harbored an extra density (purple dot). Overlay, right. [Courtesy of Falcon et al., Nature, 2019.]
7. Protein Function
What do these nefarious forms of otherwise normal proteins do to the aging brain? Seed aggregates, perhaps? January saw news of the first living cases of amyloidosis likely caused by transplanted brain tissue that presumably contained amyloid seeds (Jan news). Three adults with unusually early-onset cerebral amyloid angiopathy had received grafts of pieces of dura—the membrane under the skull that envelops the brain. Up until the year 2000, dura was used in neurosurgery, or freeze-dried as a plug for leaky blood vessels. As children, two of these CAA patients had had neurosurgery, the third a vascular repair. In their 30s and 40s, they developed seizures, and bleeds in the brain. Tissue biopsy, amyloid PET, and CSF Aβ indicated extensive CAA and some cortical plaques. The findings support the idea that tiny amounts of Aβ transferred into the brain or blood vessels can kick-start amyloidosis years later.
Separately, long-mysterious protein species—the soluble fragments of APP shed by secretases—may be closer to giving up their secrets. They appear to bind GABAB-R1a, a form of the GABAB receptor. In doing so, sAPP, particularly a 17-amino-acid fragment that comprises the GABA receptor binding site, blocks neurotransmitter release in mouse hippocampus and may even oppose Aβ, calming overactivated neurons (Jan news). The idea is that sAPP tempers neural activity.
Aβ dimers were said to hamper glutamate cleanup at synapses by slowing diffusion of glutamate receptors, prolonging synaptic activity (Aug news). How Aβ does this remains unclear.
But then: Hello again, prion protein (PrP). Work published at the close of the year suggested Aβ interacts with this previously proposed “receptor” (Corbett et al., 2019; Gomes et al., 2019). PrP may be resurfacing as a proposed nexus in neurodegeneration more broadly, since it also binds tau. In fact, tau accumulates in synapses in both AD brain and mouse models, where it conspires with Aβ to cause not only hyperactivity but decreased expression of synaptic genes—and increased expression of PrP (Pickett et al., 2019).
Tau has given up some secrets about how it alters microtubule dynamics inside the cell. It condenses in patches onto microtubules, shielding them from proteases and affecting molecular motor transport up and down their length (Sep news). These dynamic patches required a minimum level of tau, raising questions about therapeutic strategies to deplete the protein.
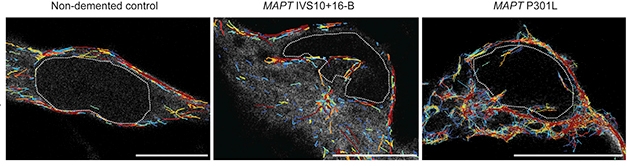
Poked. In neurons derived from healthy controls, microtubules (colored lines) skirt the nucleus (left). In neurons derived from FTD patients who carry tau mutations (middle, right), microtubules push the nuclear membrane inward, distorting the nucleus. [Courtesy of Paonessa et al., 2019.]
In contrast, mutant tau seems to compromise microtubules. In neurons derived from FTD patients with tau mutations, microtubules punched into the nuclear membrane rather than skirted it, deforming the organelle (see image above and Jan news). Prior work had suggested tau can disrupt nucleocytoplasmic transport and even scupper RNA processing (Dec 2017 news; Sep 2018 news). This year, one report claimed that mutant tau interacts with cytoskeletal components, shortening axon initial segments and causing hyperexcitation (Sep news).
8. Vascular System
From clinical trials to cell biology, the field’s focus on how changes in the cardio- and cerebrovascular system affect a person’s risk for dementia intensified in 2019. The Sprint MIND hypertension control trial results were published, and a two-year follow-up study started (Jan news). The trial had found that lowering systolic blood pressure to 120 mmHg for three years in people over 50 nudged down their subsequent rate of MCI by 20 percent. The follow-up will look for a longer-lasting effect on dementia.
On a more sobering note, the HOPE-3 trial found that neither blood pressure nor cholesterol medication slowed cognitive decline in people over 70 (Mar news). However, this trial reduced systolic blood pressure only by 6 mmHg on average, to 136, which is still slightly elevated.
More evidence surfaced that vascular risk for dementia starts shockingly early in life. In the British Birth Cohort, a group of people studied since they were born in 1947, vascular problems as early as their 30s put them at risk for dementia late in life. They are now 72 (Nov news). Importantly, their vascular risk did not correlate with brain amyloid deposition, reinforcing separate etiologies for AD and other types of dementia.
One experimental strategy for treating AD and other brain disorders drew debate. Some scientists believe that temporarily opening the blood-brain barrier might deliver larger amounts of therapeutis, such as antibodies, to the brain. In a small study, focused ultrasound opened the barrier in the cortices of five volunteers with early AD, for several hours. The breach seemed to bring few adverse effects, though neuronal connectivity on functional MRI scans was temporarily reduced (Meng et al., 2019). A larger trial is being planned. Other scientists reported at CTAD that the principle works in APP23 mice. A combination of an aducanumab analog plus scanning ultrasound restored memory better than either the antibody or ultrasound alone. Scanning ultrasound transiently opens the BBB across the brain in a wave. This helped a full-size anti-tau immunoglobulin diffuse into the brain of a mouse model of tauopathy (Janowicz et al., 2019). A safety trial will begin in early 2020.
On the other side of this coin, breaches in an aging blood-brain barrier have long been suspected to be a mechanism by which vascular disease increases risk for dementia. Exactly what is going on at the BBB, however, is controversial. In December, a leaky barrier was linked to changes in neuronal network activity, silent seizures, and cognitive loss in people with MCI/AD or epilepsy. The deficits were blamed on astrocyte TGFβ receptors activated by serum albumin, and TGFβ receptor antagonists remedied the problem—at least in mice (Dec news).
In people, BBB leaks in the hippocampus are thought to cause cognitive decline independently of Aβ, tau, or vascular pathology. In 2019, new data in this line of research pointed at damage to pericytes, specialized little cells that surround the blood vessels of the brain (Jan news). The findings suggest that the hippocampus may be particularly prone to BBB disruption.
Pericytes may also constrict blood vessels in response to Aβ, reducing blood flow (Jun news). Circulating neutrophils might do the same. In mouse models of amyloidosis, these immune cells clogged the teeny capillaries of the brain and reduced blood perfusion by as much as 30 percent. An antibody that prevents neutrophils from sticking to the vessels’ walls restored blood flow and memory (Feb news).
What about the endothelial cells that line the blood vessels and help maintain the BBB? Researchers discovered an endothelial-gene-expression signature common to stroke, seizure, brain inflammation, and trauma that might be a marker for BBB damage, though that work was in mice (Nov news).
Another line of research implicates endothelial receptors in brain aging. Prior studies using parabiosis, in which the circulatory systems of old and young mice are spliced together, indicated that something in old plasma activates microglia and inhibits neurogenesis. A 2019 continuation of that work implicated VCAM1, whereby endothelial cells on the BBB make this immunoglobulin receptor in response to injury or as the brain ages. VCAM1 reportedly attracts leukocytes on the luminal side of the barrier, which somehow activates the microglia on the brain side of the barrier; this suppresses neurogenesis (May news). Blocking VCAM1 mitigated the aging effect of old plasma on young mice, and boosted neurogenesis in old mice. Once again linking microglia to what goes down in blood, blocking the B cell receptor CD22 reprogrammed microglia in old mice to look more like those in young mice. An anti-CD22 antibody boosted phagocytosis of Aβ and other neurodegeneration proteins (Apr news). CD22 generally suppresses B cells so immune responses don’t run amok. The cell surface protein is elevated in the AD brain, where it may suppress normal microglial function.

VCAM1 Opens Door to Aging. In young mice, BECs express little VCAM1, microglia are ramified and calm, neurogenesis proceeds. In old mice, BECs ramp up VCAM1 expression, which tethers leukocytes to their luminal side. This activates microglia and suppresses neurogenesis. Anti-VCAM1 antibodies block the effects of aged plasma, rejuvenating the brain (bottom). [Courtesy of Yousef et al., Nature Medicine, 2019.]
Adult neurogenesis is a perennial topic of fascination in neurovascular research, and in 2019 there were new—but still no consensus—on whether or how neurogenesis benefits the mammalian brain. In mice, “runner plasma,” taken from animals who spun the exercise wheel and injected into sedentary mice, boosted their neurogenesis and soothed neuroinflammation (Sep news). How that happens is unknown, but running upped plasma complement inhibitors such as clusterin, variants of which affect AD risk. When people with mild cognitive impairment exercised, their clusterin rose.
Contrary to prior findings, the aging human brain apparently does continue to make new neurons, though not the AD brain (Mar news; May news). Could the dearth of new neurons in the AD brain be caused by lack of REST? This transcription factor promotes healthy aging. REST expression falls in AD and, without it, progenitors differentiate into neurons too fast, depleting the progenitor pool and ultimately exhausting neurogenesis (Feb news). Long-lived people had more REST in their neurons than people who had died younger (Oct news).
Finally, research in 2019 continued to erode the dogma that the mammalian brain is “immune privileged.” It appears that a specialized form of T cell hangs out in the meninges—the protective membranes that wrap around the parenchyma. These T cells are not wrangling microbial invaders, but they do produce copious amounts of IL-17, which boosts brain-derived neurotrophic factor, improving short-term memory (Oct news).
9. Imaging
A big step forward in 2019 came as a result of nuclear medicine's new ability to track both amyloid and tau pathology with PET over time in living people. Data from this combination further strengthened the hypothesis that amyloid pathology kicks off Alzheimer’s pathogenesis by driving the accumulation of pathologic tau, which, in turn, cripples cognition (see image below). Importantly, how fast amyloid and tau pathology grew in a person’s brain better predicted their cognitive decline years later than did their absolute levels (May news; Jun news).

Road to Ruin. Statistical modeling of serial amyloid and tau PET suggests a sequential model of decline (blue lines). Baseline Aβ three years before a tau PET scan (t=-3) drives Aβ change and tau change after the scan (t=0 to t=+2). In turn, tau change best correlates with cognitive decline six years after the initial Aβ scan (t=+3). Other scenarios explaining cognitive decline (black and dotted lines) are less likely. [©2019 Hanseeuw BJ et al., JAMA Neurology.]
Amyloid has never correlated well with cognitive decline. Many people have a high plaque burden but no obvious dementia at a single time point, and this has fed doubt about the amyloid hypothesis. Over the past decade, presymptomatic observation studies have solved this problem by painting a more dynamic picture of staged biomarker change. In 2019, researchers capitalized on this type of longitudinal data to develop the concept of amyloid chronicity, or duration. It says that it’s not so much the amount of amyloid in the brain that’s predictive, but how long the amyloid has been there (Oct news). People start accumulating amyloid at different ages, and previously scientists were unable to tell from a single scan how long a person had been amyloid-positive; chronicity, by contrast, correlates tightly with amyloid PET (see image below). Amyloid chronicity also predicted cognitive decline and accumulation of tau in the entorhinal cortex better than did amyloid load per se at one scan.

How Long You Had It. Different people start accumulating amyloid at different ages, obscuring simple amyloid load/age correlations (left). A correlation emerges between amyloid load and chronicity (right). Colors represent trajectories. [Courtesy of Sterling Johnson.]
Broadly analogous to duration of protein pathology, a typical pattern of brain aging also emerged from MRI scans that were analyzed with a new machine learning technique. It showed that the brains of people with a neurologic disease looked older than their chronological age (Oct news).

Long-Distance Relationship. Aβ in regions of the default mode network, including precuneus (green dot), posterior cingulate (yellow), inferior parietal (blue), medial prefrontal (pink), and lateral temporal (red) correlates with hypometabolism at distant but connected sites in the DMN (orange lines). Aβ in other regions, including the anterior cingulate (light blue) and paracentral cortex (orange) poorly associated with hypometabolism, and only outside the DMN. [Courtesy of Pascoal et al., Nature Communications.]
Correlations with amyloid tightene further when researchers looked at different areas of the brain, deploying multiple imaging techniques at once. For example, amyloid in some regions, such as the default-mode network, correlated with hypometabolism in other regions, indicating that amyloid affects both adjacent neurons and distant, connected neurons (Jun news; see image at right). When combined, amyloid and distal hypometabolism correlated with cognitive decline.
In frontotemporal dementia, connectivity imaging proved useful in a similar way. Baseline structural MRI scans were used to pinpoint, in a given person, where their disease had started, and that epicenter then predicted subsequent atrophy in connected regions (Oct news).
Imaging has been a huge help in differential diagnosis, allowing researchers to tell AD apart from other forms of dementia. Now, it is even being used to distinguish subtypes of Alzheimer’s. For example, hypometabolism in the frontal cortex marked a behavioral variant of AD from typical AD (Sep news). This disease is easily mistaken for FTD (see image below).

Frontal Hypometabolism. On FDG PET scans, hypometabolism in frontal areas distinguishes bvAD from typical AD, and matches areas of hypometabolism in bvFTD. [Courtesy of Singleton et al., 2019.]
As an aside on the topic of differential diagnosis, 2019 saw the introduction of a new suggested disease. Called limbic-predominant age-related TDP-43 encephalopathy, it sparked lively controversy (May news). Is LATE really a disease, or a catchy acronym for a postmortem pattern? One in five people over age 80 have this neuropathology at death, but does a distinctive symptom pattern go with it during life? This new neuropathological diagnosis is sometimes associated with dementia, but other old people who died with their cognition intact have TDP-43 in the same limbic areas.
As AD research has plunged headlong into studying inflammation and the role of microglia, imaging for both has lagged behind because specific tracers have been hard to find. Did the field finally get a PET tracer for microglia in 2019? 11C-CPPC, a ligand that binds colony stimulating factor 1 receptors, lights up areas of the brain affected by inflammation (Jan news). Only microglia and macrophages express CSF1R, making CPPC a more specific tracer than the TSPO ligands currently in use. The mitochondrial protein TSPO is expressed by microglia, astrocytes, and endothelial cells. More CPCC bound in the brains of APP transgenic mice than wild-type, and postmortem AD brain bound more CPCC than control (see image below).

New Picture of Neuroinflammation? A new PET tracer binds to three postmortem AD but not control brains (top row); unlabeled tracer blocks this uptake (bottom). [Courtesy of Horti et al., PNAS].
10. Environment/Lifestyle
Microbes
In 2019, more AD researchers became actively interested in the long-standing idea of infectious origins for neurodegenerative diseases. The year ended on sad news for investigators in this area and beyond. Just days before Christmas, Robert Moir of Massachusetts General Hospital, an intrepid, irreverent, and beloved Aussie who 10 years ago introduced the idea that Aβ is primarily an antimicrobial protein, succumbed to brain cancer at the young age of 58 (NYT editorial; Apr 2009 conference news). Moir doggedly developed his idea up until his death (Eimer et al., 2018; Moir and Tanzi, 2019), slowly persuading colleagues in the field. One broadly analogous line of research showed that the PD risk gene LRRK2 helps defend organisms against microbial infection, at the cost of increasing risk of α-synuclein aggregation (Shutinoski et al., 2019).
Separately, virologists followed up on a high-profile 2018 paper about human herpesvirus DNA in the brains of people with AD. They invited Alzheimerologists to a topical workshop at their 2019 International Conference on human herpesviruses-6 and -7. It concluded, after a day’s debate, that more work is needed (Jul conference news). Recently, biostatisticians questioned whether human herpesviruses HHV6A and HHV7 are really more abundant in AD brain (Jeong and Liu, 2019). Other groups mined large datasets in search of correlations between infection and neurodegenerative disease. One reported protection from Parkinson’s in people treated for hepatitis C infection with interferon (June news), another identified patterns of differential gene expression common to AD, PD, and viral infection (Costa et al., 2019). Even mouth flora has been blamed. Some researchers claim that the gum bacterium Porphyromonas gingivalis can enter the brain and that one of its proteases is elevated in AD brain, with a clinical trial of a protease inhibitor underway (Jan news).
Exercise
New evidence from large observational studies further cemented the well-accepted idea that physical activity protects against all-cause dementia. Some new evidence supported the less-accepted notion that exercise protects against AD specifically, as well. Among people older than 50, more physical activity associated with reduced AD biomarker and markedly less dementia over five to14 years in U.S., U.K., and Chinese cohorts (Jul conference news).
Young people may benefit, too. In a long-standing observational cohort in New York City, people who had exercised vigorously since their teenage years were 60 percent less likely to develop dementia decades later. Separately, a comparison of data from a personality survey of high school students taken in the 1960s with the same people’s current medical records linked an adolescent trait—back then called “vigor"—with low dementia incidence now. Vigor might be a proxy for physical activity (Oct news).
Researchers are starting to define how exercise protects. Data from the Harvard Aging Brain Study showed less brain atrophy and cognitive decline associated with baseline levels of brain amyloid (Rabin et al., 2019).
Sleep
Sleep research has taken a firm footing in Alzheimerology. In 2019, one sleep study made national news when it said slow-wave neural oscillations during deep sleep evoke rhythmic waves of cerebrospinal fluid washing through the brain (Nov news). This study was not in people with AD, but researchers are now looking at whether this slow-wave oscillation clears solutes such as Aβ.
Rhythms from without the brain might help, too. Rocking like a baby deepens sleep in adults, as well, extending time in non-rapid eye movement sleep and improving memory (Jan news). The non-REM sleep included more slow-wave oscillations, that is, synchronized neural rhythms of 1.5 Hz or less that can be detected by electroencephalography. In AD patients, less non-REM slow-wave activity correlated with tau tangles and to a lesser extent with amyloid plaques (Jan news).
Faster waves, in the 40 Hz range, may be important during waking hours. Researchers had reported previously that a flashing light can entrain neural activity in this gamma frequency, which reduced plaques in the mouse brain. In 2019, sound was reported to achieve the same synchronicity in the auditory cortex and hippocampus, and sound and light together reportedly caused synchronicity deep in the sensory cortex, where it stimulated microglia to remove Aβ (Mar news). Gamma entrainment using sensory stimuli (GENUS) has since been used in more models of amyloidosis and tauopathy, where it appears to curb microgliosis, plaques, and phosphorylated tau, strengthen synapses, and improve memory (May news). GENUS is being tested in a Phase 1 trial of people with prodromal or mild AD.—Tom Fagan and Gabrielle Strobel
References
Therapeutics Citations
- Aduhelm
- Leqembi
- Gantenerumab
- Kisunla
- Crenezumab
- Amilomotide
- Umibecestat
- Verubecestat
- Elenbecestat
- Tilavonemab
- Gosuranemab
- Naproxen
- Apabetalone
- AL002
- AL003
- Nuplazid
- GV-971
- Gemfibrozil
News Citations
- Biogen/Eisai Halt Phase 3 Aducanumab Trials
- ‘Reports of My Death Are Greatly Exaggerated.’ Signed, Aducanumab
- Exposure, Exposure, Exposure? At CTAD, Aducanumab Scientists Make a Case
- Amyloid Clearance: Check. Cognitive Benefit: Um … Maybe.
- Roche Pulls Plug on Two Phase 3 Trials of Crenezumab
- Results from Verubecestat APECS Trial Published
- Cognitive Decline Trips Up API Trials of BACE Inhibitor
- End of the BACE Inhibitors? Elenbecestat Trials Halted Amid Safety Concerns
- Picking Through the Rubble, Field Tries to Salvage BACE Inhibitors
- AbbVie’s Tau Antibody Flops in Progressive Supranuclear Palsy
- Anti-Tau Antibody Looks Safe, Hits Target
- Gosuranemab, Biogen’s Anti-Tau Immunotherapy, Does Not Fly for PSP
- Closing the Book on NSAIDs for Alzheimer’s Prevention
- Going Indirect: Can Therapies Halt Alzheimer’s from Outside the Brain?
- Antibodies Against Microglial Receptors TREM2 and CD33 Head to Trials
- Could CD33 Be the Microglial Target for Stimulating Phagocytosis?
- Phase 3 Trial Suggests Pimavanserin Assuages Psychosis in Dementia
- China Approves Seaweed Sugar as First New Alzheimer’s Drug in 17 Years
- Time to Try Again: Gene-Based Therapy for Neurodegeneration
- Lysosomal Diseases: Stepping Stones to Gene Therapy for Alzheimer’s?
- At CTAD, Early Failures and Hints of Success, from Small Trials
- As DIAN Wraps Up Anti-Aβ Drug Arms, it Sprouts Tau, Primary Prevention Arms
- Dementia Researchers Commend FDA Crackdown on Supplement Hype
- Paper Alerts: Massive GWAS Studies Published
- AD Genetic Risk Tied to Changes in Microglial Gene Expression
- Cell-Specific Enhancer Atlas Centers AD Risk in Microglia. Again.
- In Pathology Cascade, Microglia Rev Up After Plaques but Before Tangles
- The Mutation You Want: It Protects the Brain, Extends Life
- Are Disease Mutations Lurking Within ‘Dark Regions’ of the Genome?
- Mysterious RNA Circles Crop up in Alzheimer’s Brain
- Can an ApoE Mutation Halt Alzheimer’s Disease?
- Is APOB a Risk Gene for Early Onset Alzheimer’s?
- Genetics Propels DIAN Toward Therapies
- Familial AD Mutations, β-CTF, Spell Trouble for Endosomes
- Multi-Gene Score Predicts Cognitive Decline Independently of Brain Imaging
- Parsing How Alzheimer’s Genetic Risk Works Through Microglia
- Paper Alert: MS4A Variants May Sway Alzheimer’s Risk Via TREM2
- In Alzheimer’s, More TREM2 Is Good for You
- Without TREM2, Plaques Grow Fast in Mice, Have Less ApoE
- TREM2, Microglia Dampen Dangerous Liaisons Between Aβ and Tau
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
- Microglia in Disease: Innocent Bystanders, or Agents of Destruction?
- On The Docket at AD/PD: The Many Crimes of ApoE4
- Among AD Mutations, Only ApoE4 Seems to Hobble Microglia
- Expression, Expression, Expression—Time to Get on Board with eQTLs
- Rogue Gene Networks Track with Neurodegeneration Across Diseases
- Spatial Transcriptomics Uncovers Coordinated Cell Responses to Amyloid
- Single-Cell Profiling Maps Human Microglial Diversity, Flexibility
- The Human Brain Hosts a Menagerie of Microglia
- When It Comes to Alzheimer’s Disease, Do Human Microglia Even Give a DAM?
- Hot DAM: Specific Microglia Engulf Plaques
- Chimeric Mice: Can They Model Human Microglial Responses?
- Human Microglia Make Themselves at Home in Mouse Brain
- New Tool: Microglia-Specific Reporter Mice, With Choice of Colors
- Are Microglia Plaque Factories?
- In Tauopathy, ApoE Destroys Neurons Via Microglia
- ApoE4 and Tau in Alzheimer’s: Worse Than We Thought? Especially in Women
- Is a Woman’s Brain More Susceptible to Tau Pathology?
- Do Tribes of Astrocytes Wage War on Synapses?
- Dopaminergic Neurons Conjured from Astrocytes Restore Motion
- Are Aβ Blood Tests Ready for Prime Time?
- Drawing Closer: Alzheimer’s Blood Test for Primary Care
- Why Bother With Round Robins on Blood Tests? Q&A with Kaj Blennow
- Move Over Aβ, CSF P-Tau Tells Us There’s Plaque in the Brain
- Move Over CSF, P-Tau in Blood Also Tells Us There’s Plaque in the Brain
- Neurofilament in Blood Foretells Early Onset Alzheimer’s
- Proteomics Uncovers Potential Markers, Subtypes of Alzheimer’s
- Fluid AD Biomarkers Link P-Tau to Synapses, Inflammation
- Synaptic Proteins in CSF: New Markers of Cognitive Decline?
- Do Brain Waves During Sleep Reflect Aβ and Tau Pathologies?
- Right Turn: Aβ Fibril Structure from Alzheimer’s Brain Reveals Surprising Twist
- CryoEM γ-Secretase Structures Nail APP, Notch Binding
- BACE and γ-Secretase Form Mega-Complex that Processes APP
- Conformers Confirmed: Structure of Pick’s Tau Distinct from AD Tau
- Traumatic Tau: Filaments from CTE Share Distinct Structure
- First In Vivo Look at Amyloidosis Sparked by Dural Grafts
- Secreted APP Binds GABA-B Receptors, Blocks Neurotransmitter Release
- Aβ Dimers Block Glutamate Uptake, Fire Up Synapses
- Islands of Tau Coat and Protect Cytoskeleton
- Invasion of the Microtubules: Mutant Tau Deforms Neuronal Nuclei
- Is There No End to Tau’s Toxic Tricks?
- Tau Stymies Transport Through Neuron’s Nucleus
- Mutant Tau Stiffens Axon Cytoskeleton Near Soma
- SPRINT MIND Data Published, Follow-Up Extended
- Too Little, Too Late? Blood Pressure and Cholesterol Meds Don’t Slow Cognitive Decline in 70s
- Already in Mid-30s, Poor Vascular Health Means Small Brain at 70
- Does a Breached Blood-Brain Barrier Cause Seizures in AD?
- Absent Aβ, Blood-Brain Barrier Breakdown Predicts Cognitive Impairment
- Aβ Acts Through Pericytes to Throttle Brain Blood Flow
- Immune Cells Clog Capillaries in Mice, Disrupt Memory
- Scientists Discover a Common Distress Signal in the Blood-Brain Barrier
- Paper Alert: VCAM1 Opens the Door to Brain Aging
- CD22 Suppresses Microglial Phagocytosis—A New Therapeutic Target?
- 'Runner Plasma' Jogs Neurogenesis, Quells Neuroinflammation in Mice
- Neurogenesis Tapers In Older Brains, But Plummets in Alzheimer’s
- Neurogenesis Wanes in Alzheimer’s, Along With Cognition
- In Alzheimer’s, Too Little REST Spurs Too Much Neurogenesis
- Could Getting Enough REST Extend Your Life?
- Do Immune Cells in the Meninges Help with … Memory?
- Longitudinal Tau PET Links Aβ to Subsequent Rise in Cortical Tau
- Serial PET Nails It: Preclinical AD Means Amyloid, Tau, then Cognitive Decline
- Amyloid—It’s Not Whether, but for How Long You’ve Had It
- MRI and Machine Learning Depict Brain Aging in Health, Disease
- Parsing Local and Distal Aβ Shows Links to Metabolism, Cognition
- Imaging Model Locates Epicenter of Disease, Predicts Atrophy
- Frontal Metabolic Patterns Mark Behavioral Subtype of AD
- Introducing LATE—A Common TDP-43 Proteinopathy that Strikes After 80
- New PET Tracer Selectively Lights Up Activated Microglia in Brain
- Prague: Aβ Rehabilitated as an Antimicrobial Protein?
- Going Viral: Alzheimer’s Research at Herpes Conference
- Study Strengthens Link between Hepatitis C Virus, Parkinson’s
- An Antimicrobial Approach to Treating Alzheimer’s?
- Physical Activity May Shield the Brain from the Onslaught of Aβ
- Does Your Personality in High School Foretell Your Dementia Risk?
- Deep Sleep Makes Waves for CSF
- Rocking Improves Sleep and Memory in Adults
- Tau, More than Aβ, Affects Sleep Early in Alzheimer’s
- Flash! Beep! Gamma Waves Stimulate Microglia, Memory
- Gamma Waves Synchronized by Light: Good for Synapses, Memory?
Mutations Citations
Research Models Citations
Paper Citations
- Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Larson A, Pendergast MK, Goldkind SF, Lee EA, Kuniholm A, Soucy A, Vaze J, Belur NR, Fredriksen K, Stojkovska I, Tsytsykova A, Armant M, DiDonato RL, Choi J, Cornelissen L, Pereira LM, Augustine EF, Genetti CA, Dies K, Barton B, Williams L, Goodlett BD, Riley BL, Pasternak A, Berry ER, Pflock KA, Chu S, Reed C, Tyndall K, Agrawal PB, Beggs AH, Grant PE, Urion DK, Snyder RO, Waisbren SE, Poduri A, Park PJ, Patterson A, Biffi A, Mazzulli JR, Bodamer O, Berde CB, Yu TW. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N Engl J Med. 2019 Oct 24;381(17):1644-1652. Epub 2019 Oct 9 PubMed.
- Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee SH, Haddick PC, Ngu H, Modrusan Z, Larson JL, Kaminker JS, van der Brug MP, Hansen DV. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer's Disease Not Evident in Mouse Models. Cell Rep. 2018 Jan 16;22(3):832-847. PubMed.
- Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, Chen Y, Su Y, Myers AJ, Hardy J, Paul Vonsattel J, Younkin SG, Bennett DA, De Jager PL, Larson EB, Crane PK, Keene CD, Kamboh MI, Kofler JK, Duque L, Gilbert JR, Gwirtsman HE, Buxbaum JD, Dickson DW, Frosch MP, Ghetti BF, Lunetta KL, Wang LS, Hyman BT, Kukull WA, Foroud T, Haines JL, Mayeux RP, Pericak-Vance MA, Schneider JA, Trojanowski JQ, Farrer LA, Schellenberg GD, Beecham GW, Montine TJ, Jun GR, Alzheimer’s Disease Genetics Consortium. Exceptionally low likelihood of Alzheimer's dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020 Feb 3;11(1):667. PubMed.
- Zhou X, Chen Y, Mok KY, Kwok TC, Mok VC, Guo Q, Ip FC, Chen Y, Mullapudi N, Alzheimer’s Disease Neuroimaging Initiative, Giusti-Rodríguez P, Sullivan PF, Hardy J, Fu AK, Li Y, Ip NY. Non-coding variability at the APOE locus contributes to the Alzheimer's risk. Nat Commun. 2019 Jul 25;10(1):3310. PubMed.
- Campion D, Charbonnier C, Nicolas G. SORL1 genetic variants and Alzheimer disease risk: a literature review and meta-analysis of sequencing data. Acta Neuropathol. 2019 Aug;138(2):173-186. Epub 2019 Mar 25 PubMed.
- Olah M, Menon V, Habib N, Taga M, Yung C, Cimpean M, Khairalla A, Dionne D, Hopp S, Frosch MP, Hyman BT, Beach TG, Sarkis R, Cosgrove GR, Helgager J, Golden JA, Pennell PB, Schneider JA, Bennett DA, Regev A, Elyaman W, Bradshaw EM, De Jager PL. A single cell-based atlas of human microglial states reveals associations with neurological disorders and histopathological features of the aging brain. bioRχiv. June 11, 2018 BioRxiv.
- Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009 Nov;12(11):1361-3. PubMed.
- Mielke MM, Hagen CE, Xu J, Chai X, Vemuri P, Lowe VJ, Airey DC, Knopman DS, Roberts RO, Machulda MM, Jack CR Jr, Petersen RC, Dage JL. Plasma phospho-tau181 increases with Alzheimer's disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimers Dement. 2018 Aug;14(8):989-997. Epub 2018 Apr 5 PubMed.
- Corbett GT, Wang Z, Hong W, Colom-Cadena M, Rose J, Liao M, Asfaw A, Hall TC, Ding L, DeSousa A, Frosch MP, Collinge J, Harris DA, Perkinton MS, Spires-Jones TL, Young-Pearse TL, Billinton A, Walsh DM. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 2020 Mar;139(3):503-526. Epub 2019 Dec 18 PubMed.
- Gomes LA, Hipp SA, Rijal Upadhaya A, Balakrishnan K, Ospitalieri S, Koper MJ, Largo-Barrientos P, Uytterhoeven V, Reichwald J, Rabe S, Vandenberghe R, von Arnim CA, Tousseyn T, Feederle R, Giudici C, Willem M, Staufenbiel M, Thal DR. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019 Dec;138(6):913-941. Epub 2019 Aug 14 PubMed.
- Pickett EK, Herrmann AG, McQueen J, Abt K, Dando O, Tulloch J, Jain P, Dunnett S, Sohrabi S, Fjeldstad MP, Calkin W, Murison L, Jackson RJ, Tzioras M, Stevenson A, d'Orange M, Hooley M, Davies C, Colom-Cadena M, Anton-Fernandez A, King D, Oren I, Rose J, McKenzie CA, Allison E, Smith C, Hardt O, Henstridge CM, Hardingham GE, Spires-Jones TL. Amyloid Beta and Tau Cooperate to Cause Reversible Behavioral and Transcriptional Deficits in a Model of Alzheimer's Disease. Cell Rep. 2019 Dec 10;29(11):3592-3604.e5. PubMed.
- Meng Y, MacIntosh BJ, Shirzadi Z, Kiss A, Bethune A, Heyn C, Mithani K, Hamani C, Black SE, Hynynen K, Lipsman N. Resting state functional connectivity changes after MR-guided focused ultrasound mediated blood-brain barrier opening in patients with Alzheimer's disease. Neuroimage. 2019 Oct 15;200:275-280. Epub 2019 Jun 26 PubMed.
- Janowicz PW, Leinenga G, Götz J, Nisbet RM. Ultrasound-mediated blood-brain barrier opening enhances delivery of therapeutically relevant formats of a tau-specific antibody. Sci Rep. 2019 Jun 25;9(1):9255. PubMed.
- Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, György B, Breakefield XO, Tanzi RE, Moir RD. Alzheimer's Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron. 2018 Jul 11;99(1):56-63.e3. PubMed.
- Moir RD, Tanzi RE. Low Evolutionary Selection Pressure in Senescence Does Not Explain the Persistence of Aβ in the Vertebrate Genome. Front Aging Neurosci. 2019;11:70. Epub 2019 Mar 28 PubMed.
- Shutinoski B, Hakimi M, Harmsen IE, Lunn M, Rocha J, Lengacher N, Zhou YY, Khan J, Nguyen A, Hake-Volling Q, El-Kodsi D, Li J, Alikashani A, Beauchamp C, Majithia J, Coombs K, Shimshek D, Marcogliese PC, Park DS, Rioux JD, Philpott DJ, Woulfe JM, Hayley S, Sad S, Tomlinson JJ, Brown EG, Schlossmacher MG. Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex-dependent manner. Sci Transl Med. 2019 Sep 25;11(511) PubMed.
- Jeong HH, Liu Z. Are HHV-6A and HHV-7 Really More Abundant in Alzheimer's Disease?. Neuron. 2019 Dec 18;104(6):1034-1035. PubMed.
- Costa Sa AC, Madsen H, Brown JR. Shared Molecular Signatures Across Neurodegenerative Diseases and Herpes Virus Infections Highlights Potential Mechanisms for Maladaptive Innate Immune Responses. Sci Rep. 2019 Jun 19;9(1):8795. PubMed.
- Rabin JS, Klein H, Kirn DR, Schultz AP, Yang HS, Hampton O, Jiang S, Buckley RF, Viswanathan A, Hedden T, Pruzin J, Yau WW, Guzmán-Vélez E, Quiroz YT, Properzi M, Marshall GA, Rentz DM, Johnson KA, Sperling RA, Chhatwal JP. Associations of Physical Activity and β-Amyloid With Longitudinal Cognition and Neurodegeneration in Clinically Normal Older Adults. JAMA Neurol. 2019 Jul 16; PubMed.
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
Alector
Great review article, thank you for writing.
A few clarifying points:
1) The therapeutic goal of harnessing microglia to treat AD through activating Trem2 or inhibiting CD33 goes far beyond "spur microglia to clear amyloid." Microglia have a myriad of homeostatic and disease-fighting functions. They contain the damage from misfolded proteins, clear membrane, and cell debris, fight bacterial and viral infections, block leakage from capillaries, control the number of synapses and control the activity and/or viability of astrocytes, oligodendrocytes, endothelial cells, and nerve cells. Thus rejuvenating aged microglia to counteract AD pathology is anticipated to have broad therapeutic effects on multiple aspects of the disease.
2) The findings that "Polymorphisms associated with more CSF sTREM2 decrease a person’s chances of AD" does not mean that "TREM2 shedding is protective." The more likely explanation is that more sTrem2 is just a byproduct of more active microglia with more membrane Trem2 that are better able to counteract the disease. More membrane Trem2 will lead by default to more sTrem2 as a fixed percentage of the receptor is constantly cleaved. Several Trem2 mutations that increase the risk of AD, for example, H157Y and R47H, lead to higher levels of sTrem2. Thus the notion that sTrem2 is protective independent of the signaling receptor is not supported by human genetics and in my view, speculative.
Make a Comment
To make a comment you must login or register.